

[TOC]
代谢组学的数据分析包括:对检测到的原始数据进行数据预处理、数据质控、代谢物定量分析、生信数据挖掘等。
1.数据预处理
软件解析测序的色谱、质谱得到的图谱转变为可进行计算的数据文件,就得到了原始数据,一般的解析软件包括:Progenesis QI、MassProfiler Professional (MPP)、Sieve、MetaboAnalyst、Simca-p(代谢组学中最常用的统计分析软件)
如何使用上述的软件这里就不一一赘述了,网上有很多教程。
为了消除存在的外在因素干扰或人为实验因素影响,预处理数据可以增加数据的稳定性,常见的预处理数据步骤主要包括:
- 降噪:通过匹配滤波和移动窗平均滤波等方法,去除掉数据中来自样品制备或仪器产生的背景噪音。
- 基线校准:当没有物质时,获得的谱图强度应该为0,所有的测得的谱图强度都应以此为基准,因此需要调零校准,可以通过将同样一谱图的所有数据值减去最小值,来让基准线为0.
- 解卷积:由于物质的保留时间太接近,或者峰的宽度过大,导致多个色谱的峰没有分离而共流出时,代表多个物质的峰就会重叠或交叉在一起。解卷积,就是利用数学计算方法将色谱中未能锋利的组分重新区分。
- 峰对齐:为了校准由于仪器不稳定、样品pH和浓度等因素带来的时间偏移,需要进行峰的对齐,让所有样本中代表同一物质的谱峰保留时间一致。
- 峰识别:确定峰的起点和终点。
- 峰的特征提取:提取峰高或计算峰下的覆盖面积。
- 归一化处理:为了使不同浓度的样品之间能够具有可比性,我们需要对数据进行峰面积的归一化处理,常用的处理方法有Ctr(Center scaling),UV(unit variance scaling)和Par(Pareto scaling)。 Ctr也叫中心化,将原始数据减去每列变量的均值;UV是数据Ctr化之后,除以列变量的标准差(SD);Par是数据Ctr后处理列变量标准差的算术平方根。 Ctr将原始数据转化成离原点更近的新数据,可调节代谢物的高低浓度差异;UV的优势是所有的变量都拥有同样的权重,等同于相当的地位,缺点是某些检测误差可能会被放大;Par相比于UV则更接近院士测量数据,缺点是对变化倍数大的变量会更加敏感,容易造成不必要的变换结果。

2.数据的质控(QC)
样品较多时,我们想要获得可靠且高质量的代谢组学数据,则需要在检测时利用QC样本进行质控;QC样本时所有待测样品等量的混合物,不过若是临床样本,由于数量较多,采样时间长,则可以挑选代表性样品制备合适的QC。每间隔一定样品,添加一组QC,这样就能提供数据重复性的有效评价标准。
理论上所有的QC样本要都是相同的,不过在样品提取、监测分析过程会产生系统的误差,导致QC之间也会有差异,如果RSD<30%的特征峰,比例能达到70%以上,则说明数据良好,差异越小说明方法稳定性越高,数据质量也就越好,这体现在PCA分析图上,也就是QC样本越密集,数据越可靠。
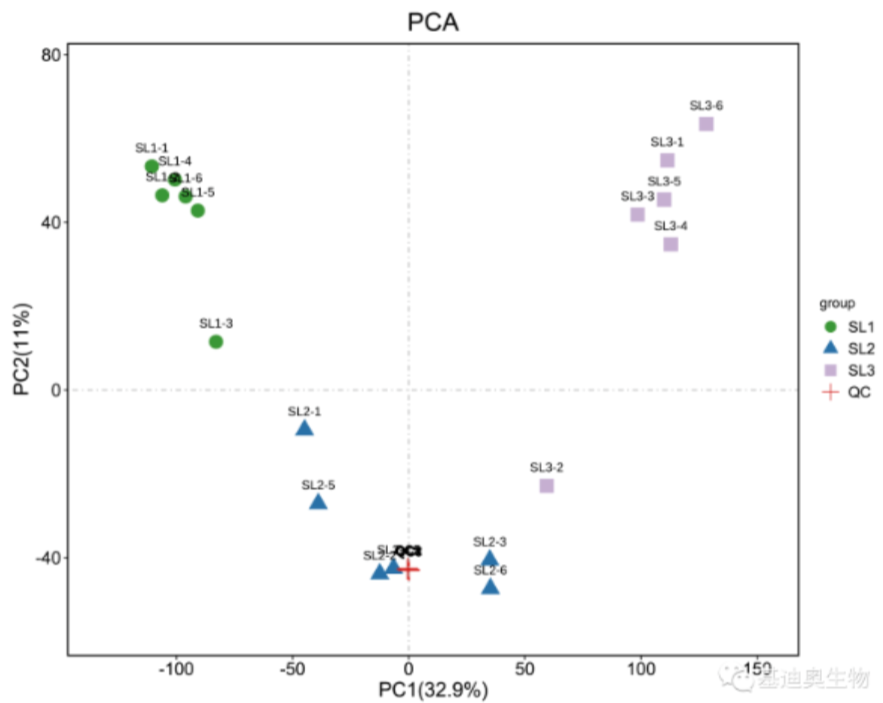
3.代谢物的定性和定量
代谢物的定性:GC-MS数据库成熟,数据量足够,一级质谱可定性,即了解化合物的分子量;二级谱图可以对化合物的结构进行分析。我们将GC质谱图与NIST、GMD数据库进行比对,将LC-MS的二级谱图与Metlin、HMDB、MzCloud等数据库或者实验室自建的数据库中的谱图进行匹配比对,就可以的搭配化合物具体的注释信息。
代谢物的定量:GC-MS他一般时通过化合物色谱峰面积来对物质进行定量的,当遇到分离度不好,色谱峰形不好,比如严重拖尾的时候,峰的面积测定就会引起很大的误差,此时可考虑使用峰高法进行定量。峰高法,顾名思义通过测量色谱图中某个组分峰的高度来对该组分进行定量。峰高法的基本原理是假设峰高(或峰面积)与组分的量成正比。具体来说,峰高法涉及以下步骤:
- 选择参考峰:首先,需要选择一个已知浓度的标准品作为参考峰。这个标准品应该是纯净的,并且浓度已知。
- 建立标准曲线:通过分析不同浓度的标准品,可以得到一系列峰高与浓度对应的数据。利用这些数据,可以绘制出标准曲线。标准曲线通常是一条直线,表明峰高(或峰面积)与浓度之间的关系是线性的。
- 测量样品峰高:在分析实际样品时,测量与标准品相对应的组分的峰高。
- 定量分析:将样品峰高与标准曲线进行比较,根据标准曲线可以确定样品中组分的浓度。
峰高法的优点是操作简单,不需要复杂的仪器设备。但是,这种方法假设色谱峰的形状和宽度在分析过程中保持不变,这在实际操作中可能并不总是成立。此外,峰高法对基线的波动比较敏感,因此对于基线不稳定的色谱图,定量结果可能会有较大误差。
LC-MS/MS利用一级质谱得到的木例子峰面积进行定量。非靶通常选择内标法进行相对定量。将内标物质,定量的加到样品中,根据预测组分和内标峰的面积或者峰高的比值进行相对定量分析。
4.代谢组学的生信分析
4.1 聚类分析
聚类分析一般有两个作用,包括:通过全部代谢物的表达谱对样本进行聚类,分析样本重复性的好坏以及分组情况;或者把表达模式相近的代谢物进行聚类,去大致的观察代谢物丰度在各组中的变化规律,常用图形包括聚类树状图和热图。
- 主成分分析(PCA): PCA是一种广泛使用的数据降维算法,它通过找出数据矩阵的方差最大方向作为第一主成分(PC1),然后在与PC1正交的平面中找出方差最大的方向作为第二主成分(PC2),以此类推。这样建立的低维平面或空间有助于分析和概览整个数据集,揭示数据集中的分组、趋势以及离群值 。
- 偏最小二乘判别分析(PLS-DA): PLS-DA结合了降维和回归模型,通过多元线性回归技术找到数据集和类别数据集之间的最大协方差方向,建立两个独立数据集的潜在关联分析方法。
- 正交偏最小二乘判别分析(OPLS-DA): OPLS-DA是PLS-DA的改进版本,它根据数据集Y的差异将数据集X的差异分为两个部分,一部分代表与Y相关的差异,另一部分代表与Y不相关的差异。OPLS-DA可以更好地区分组间差异,提高模型的有效性和解析能力。
- 差异倍数分析: 这是一种简单的分析方法,用于快速考察各个代谢物在不同组别之间的含量变化大小。它通过计算实验组与对照组的含量比值(Fold Change,FC)来实现。
- 显著性检验: 这包括t-test、方差分析(ANOVA)等方法,用于区分变量是否具有统计显著性。在代谢组学中,由于变量较多,常需要进行多重假设检验并对p值进行校正,以减少Ⅰ类错误和假阳性。
- 韦恩图: 也称文氏图,用于反映不同集合之间的交集和并集情况。在代谢组学中,韦恩图通常用于展示不同比较组之间的共有和独特的差异代谢物。
- 箱线图: 箱线图用于展示不同组间代谢物含量的分布情况。在R语言中,常用的包如ggplot2和ggpubr可用于绘制箱线图,并可以添加样本散点、坐标标题以及p值等统计信息 。
数据集通常为一个N × K的矩阵(X矩阵),N表示N个样本数,每一行代表一个样品, K表示K个变量,每一列代表一个变量,在代谢组学中变量通常是指代谢物含量。最常用的分析方法如图所示:
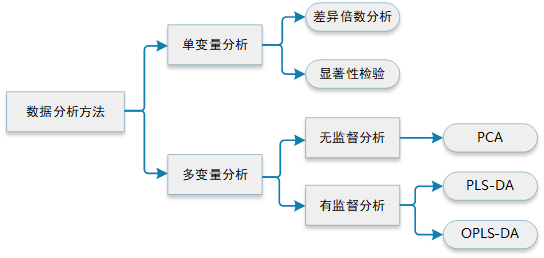

4.2 多元统计分析
代谢组学数据具有多为且某些变量间高度相关,运用传统的单变量分析无法快速、充分、准确地挖掘数据内潜在的信息。 因此对采集的多维数据,一般进行将为和归类分析(聚类),来挖掘和提炼最有用的信息。
常用的方法包括:无监督学习的PCA分析,有监督的PLS-DA分析、在PLS-DA基础上,可以加入正交信号矫正的有监督性OPLS-DA分析。

4.3 差异分析
代谢组学分析中,除了通常用到的Student’s t检验、ANOVA检验来获得P值筛选不同比较组间的差异代谢物,通常还结合多元统计分析PL-DA或者OPLS-DA的VIP值来进行筛选。也就是差异代谢物的阈值条件:OPLS-DA模型中 VIP≥1,且Student T test的P<0.05. 常用的图形包括差异柱状图、火山图、VIP图、z-score、热图和ROC图等,进行必要的数据展示。
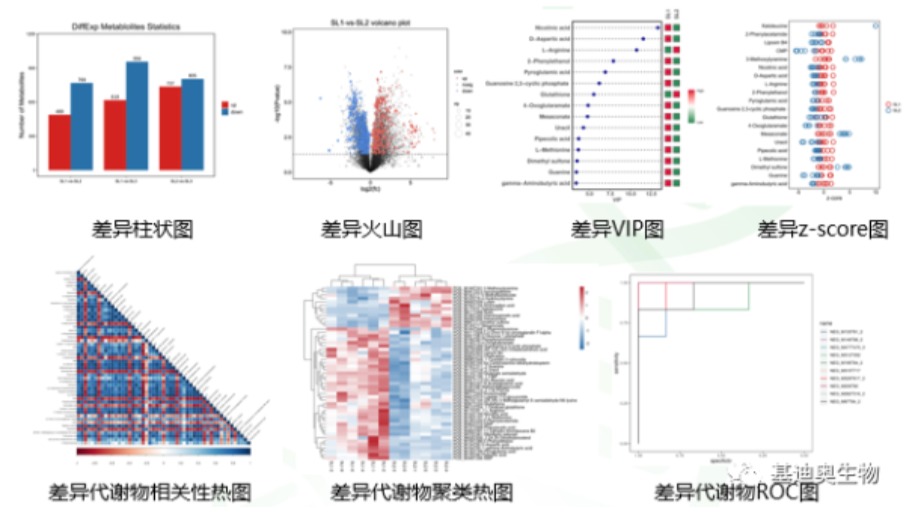
4.4 差异KEGG富集分析
在生物体内,不同的基因之间,相互协调来行使生物学功能,基于Pathway的分析有助于更进一步的了解基因的生物学功能和生物学研究的意义,甚至为临床的诊断和治疗提供一些思路、方法和理论基础。
KEGG是有关pathway的主要数据库,pathway显著性富集分析,应用超几何检验分析,找出与整个背景(基因、蛋白谱、QC样本等)相比,在差异代谢物中显著富集的pathway,从而确定差异代谢物残余的最主要生化代谢途径和信号转导方式。
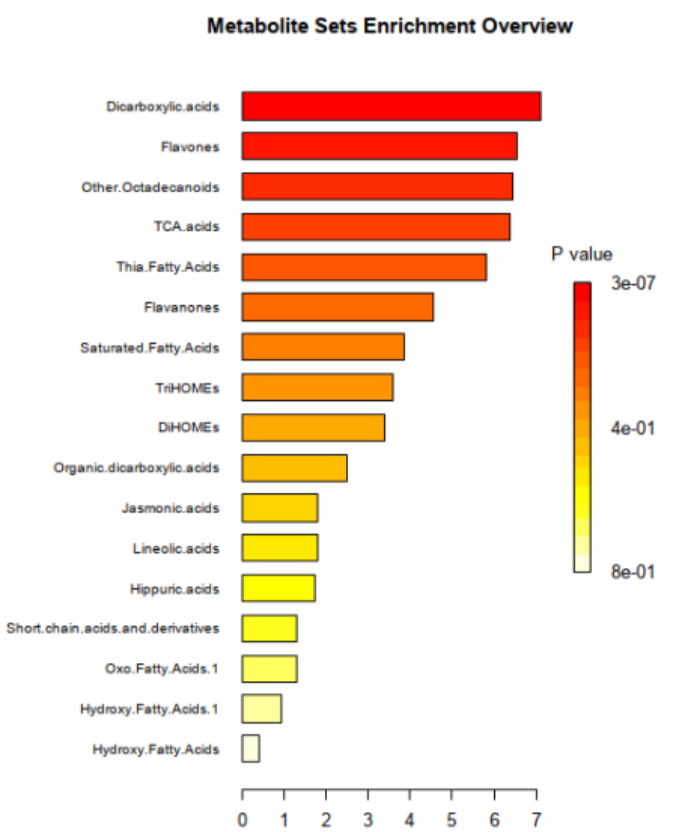
常见的图形包括:富集柱状图、富集通路图、富集热图、富集圈图、富集差异气泡图等。

传统的KEGG富集分析如果没有找到与研究相关的代谢通路,可以利用MSEA分析(Metabolite Set Enrichment Analysis)。 MSEA的QEA pattern类似于基因分析中的GSEA富集分析,主要利用The Small Molecule Pathway Database(SMPDB)数据库来进行对所有样本中鉴定到的代谢物的确定,以及解释一些重要的生物学通路中,代谢物的变化模式。
4.5 趋势分析
趋势分析,是针对多个连续样本(至少三个)的特征(样本间包含特定的时间、空间或者处理剂量的大小顺序)来对代谢物的表达模式(多阶段中表达曲线的形状)进行聚类。通过趋势分析可以找到和可视化代谢物在连续变化的组中丰度的变化趋势。
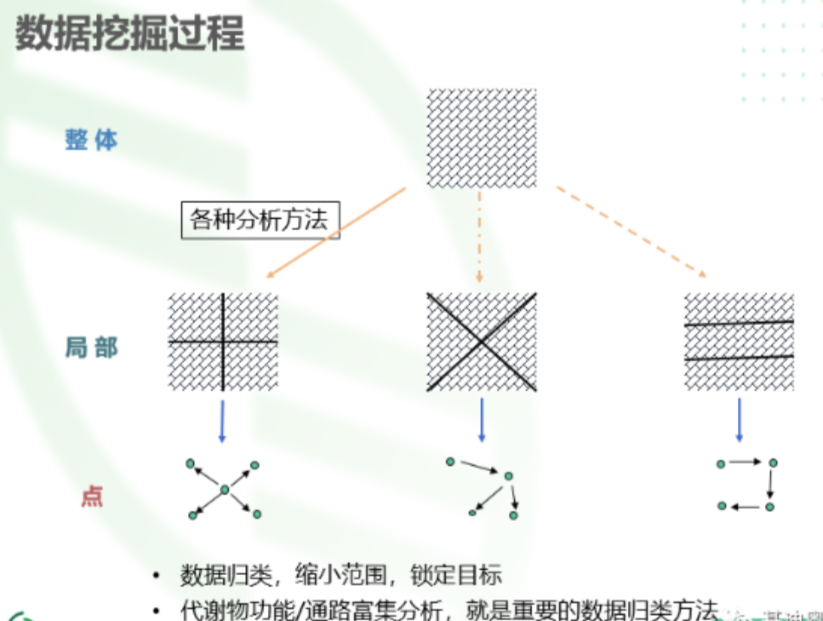
注:基于生信分析的组学数据挖掘思路,目前我了解的基本可以分成三步走,简单点就是俯瞰-拉近-聚焦,也就是整体到局部再到某个单一的点的过程。 以测序得到的海量数据作为起点,不断根据研究目标,引导和矫正数据挖掘的方向,最后确定对研究目标其重要调控的基因、蛋白、代谢物、小分子等。
代谢组学,借助差异分析、趋势分析、WGCNA分析等等,完成海量数据挖掘的第一步,完成了俯瞰到拉近的转换,获得目标代谢物集合。需要思考的是,这些代谢物具备哪些调控功能?能影响哪些生物学途径(拉近到聚焦的过程)。
5.常见代谢组学富集分析
富集分析常见的就是KEGG、MetPA和MSEA了。
三种分析策略都是以代谢物的定性定量表作为起始点,无论是自己平台还是公司的质谱检测,都可以得到这个数据表,表格中需要包含:代谢物名称、在不同样本中的风度信息等。 以此为基础借助富集分析和其他的一些分析手段找到目标代谢途径和目标代谢物。
5.1 KEGG富集
特定实验背景下,代谢物的丰度变化并非随机,往往会为了响应实验处理呈现出特定的变化趋势。 例如,动物受疾病感染后,脂类代谢、氨基酸代谢、胆汁酸代谢过程失调; 植物生长、变色过程中,花青素、类胡萝卜素、类黄酮等有机物的合成过程中,相关代谢物会随着发育成熟逐渐上调累积。
以此作为出发点,我们在特定的取样策略下,通过分析目标代谢物(包括差异代谢物、目标变化趋势代谢物、目标模块代谢物等)所在的pathway,来了解实验处理影响哪些代谢途径和内部的一些代谢物的调控方式,再结合研究目的锁定重要的代谢物。
5.1.1 KEGG分析原理
借助**超几何检验(Hypergeometric test)**判断: 质谱检测的有KEGG注释信息的代谢物(背景代谢集)在某个pathway中的代谢物数目占背景代谢集的比例,是否显著大于目标代谢集中有KEGG注释信息的代谢物(前景代谢集)在某个pathway中的代谢物数目的比例。
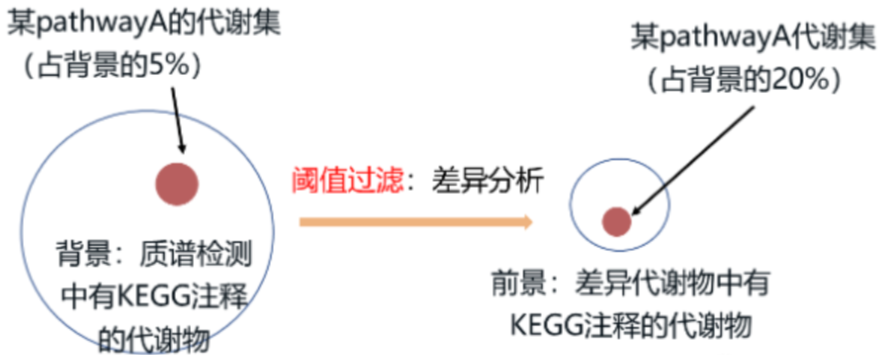
简单来说,其实就是上图中判断20%是否显著大于5%,根据得到的P value和FDR value判断前景代谢集主要参与的代谢途径。当P value或FDR value小于0.05时,则为显著富集的代谢途径。说明相比于其它途径而言,显著富集的代谢途径更易响应实验处理,出现异常调控。
对于KEGG富集分析,除了要有质谱检测的全部代谢物外,还要先有一个前景代谢集(又称目标代谢集,如图右侧的差异代谢集)。前景代谢集可通过前面说到的差异分析、趋势分析、WGCNA分析获得。
[^注]: 质谱检测得到的Name是代谢物的名称并非代谢物的KEGG注释结果,代谢物KEGG注释完成后用C_ID表示, 这个比较像基因的Symbol和ID号。代谢物开展KEGG富集分析时,需要先完成KEGG注释。
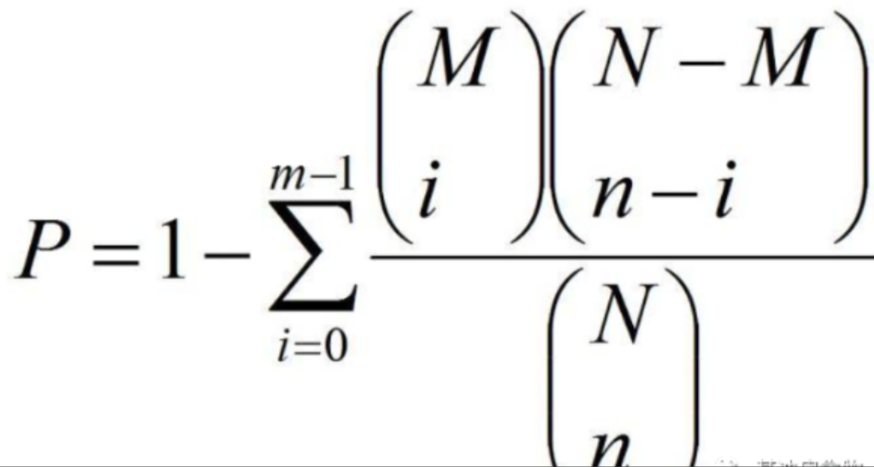
超几何检验公式
[^注]: 公式中N为质谱检测得到的所有代谢物中有KEGG注释的代谢物数目; n为N中差异代谢物数目;M为所有代谢物中注释为某特定pathway的代谢物数目;m为注释为某特定pathway的差异表达代谢物数目。
5.1.2 KEGG富集分析实现方法
这里放一个基因和蛋白的富集方法,其他同理。
进入Metascape主页(www.metascape.org),将候选基因直接复制粘贴到gene list输入框中(Step1),点击Submit按钮上传基因列表。
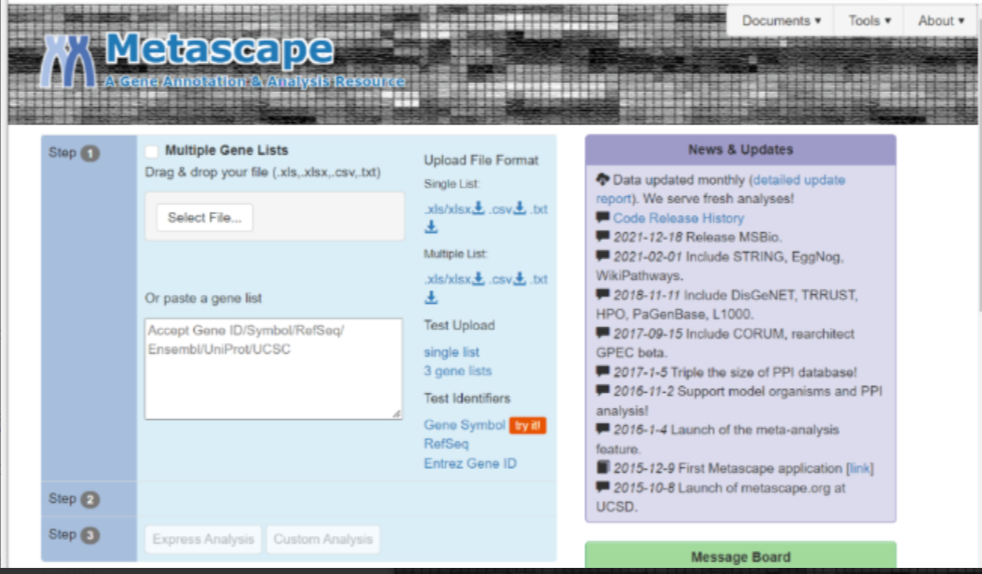
MetaScape支持的GeneID主要有Symbol、RefSeq、Ensembvl、Unipro、UCSC五种类型,下面使用Symbol进行演示:
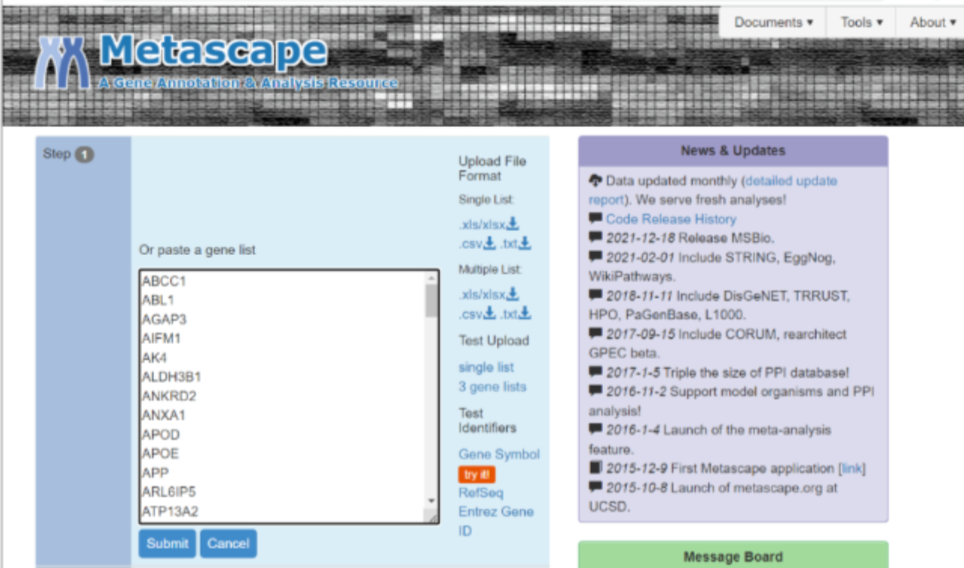
然后,在step2中选择对应的物种,我们这里选择人(H.sapiens),点击Custom Analysis进行自定义分析。
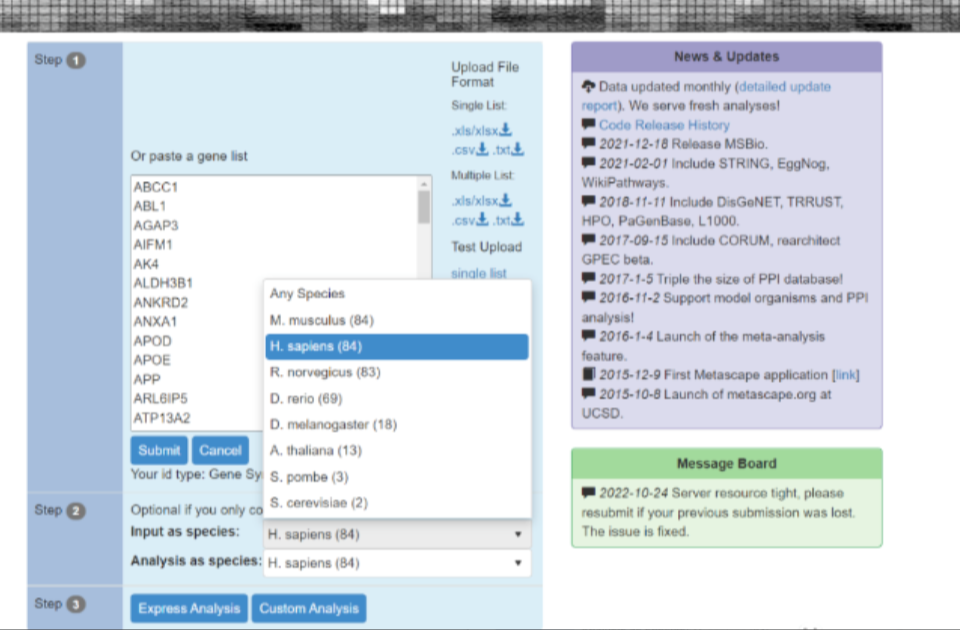
在弹出的页面中,直接选择第4个选项(Enrichment),仅勾选KEGG Pathway,取消勾选其他的数据来源,如下,然后勾选“跳过蛋白互作分析”
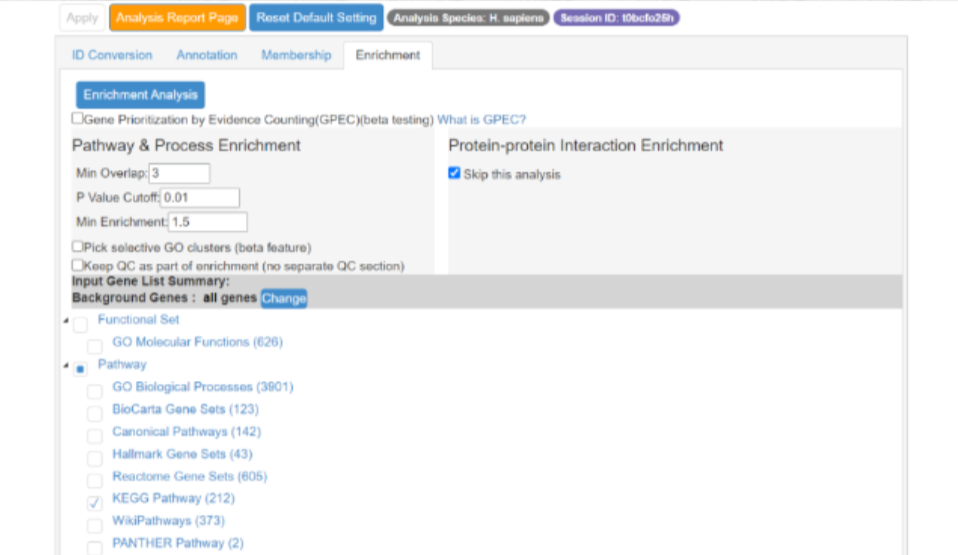
然后继续点击Enrichment Analysis蓝色按钮进行富集分析。
分析完成之后,快速浏览富集的结果,之后点击Analysis Report Page,进入分析报告页面。
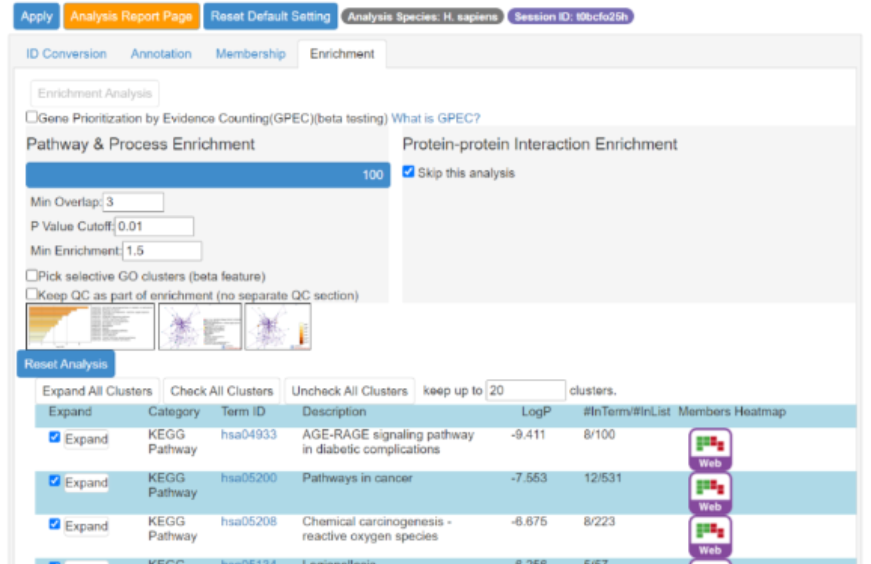
在分析报告页面中,点击Gene List Report Excel Sheets按钮,下载分析结果表格。
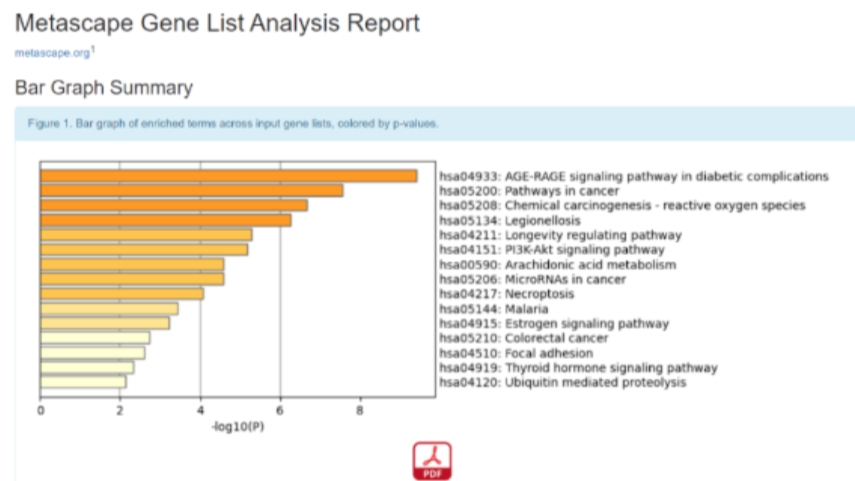
下载的分析结果表格如下,用于作图的数据主要在sheet2表格中。
完成KEGG富集分析之后,接下来我们使用ggplot2包对分析结果进行可视化!
1 | #载入相关的R包; |

1 | #提取用于作图的列; |

1 | #转成因子,防止重新排序 |

1 | #建立数据与图形或者点之间的映射关系,确定点的(x,y)坐标,绘制散点图 |
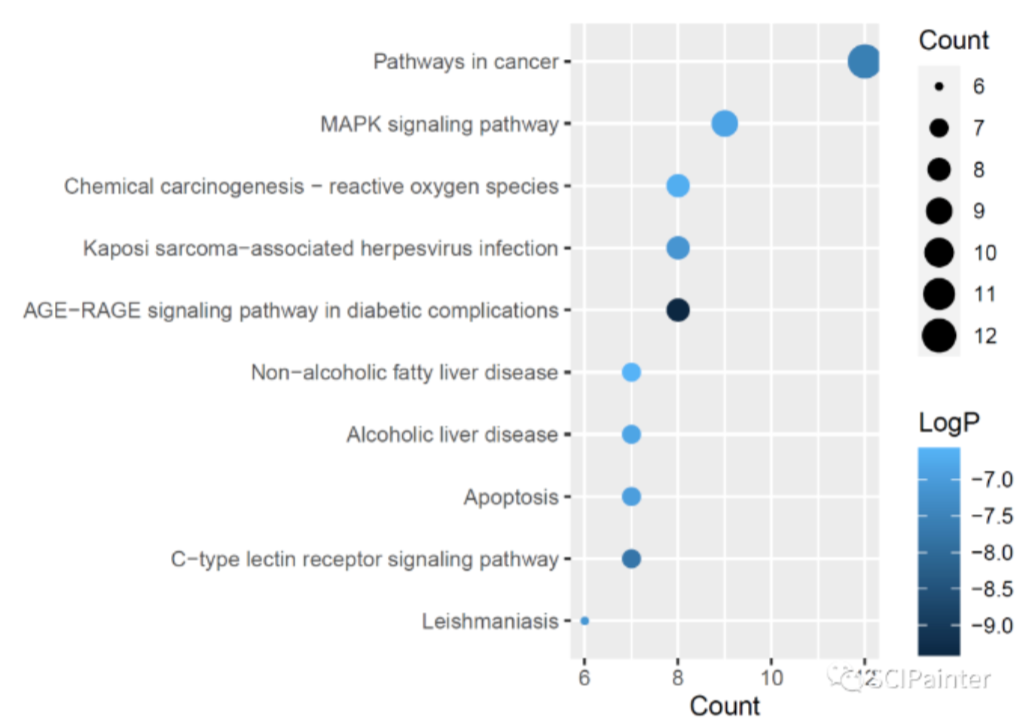
1 | #自定义颜色渐变 |
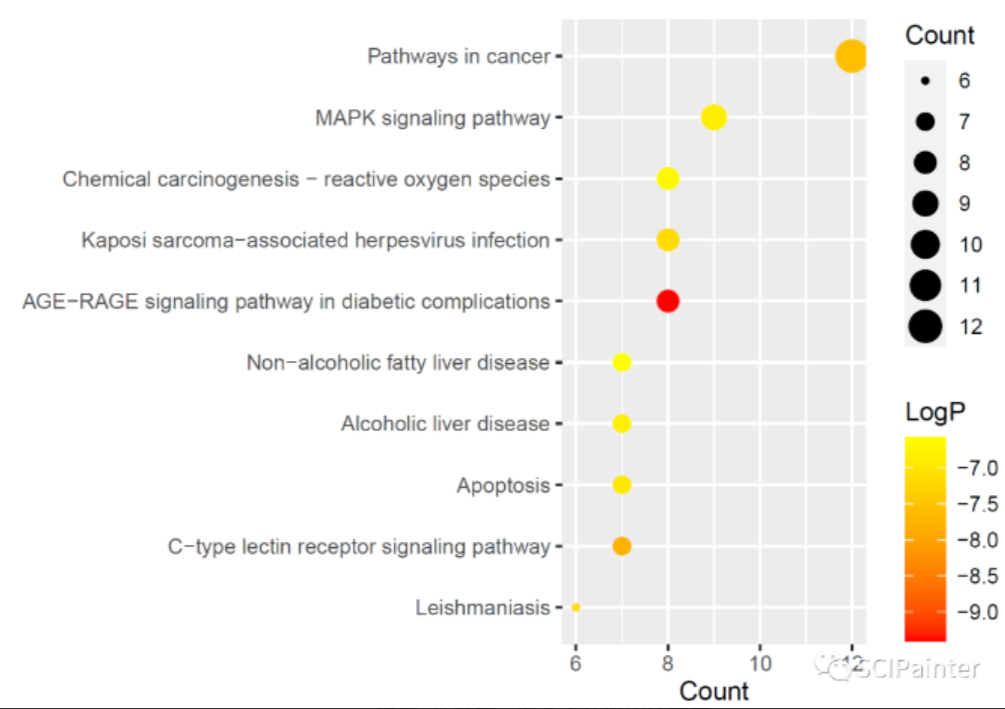
1 | #设置x轴范围,避免点溢出绘图区 |
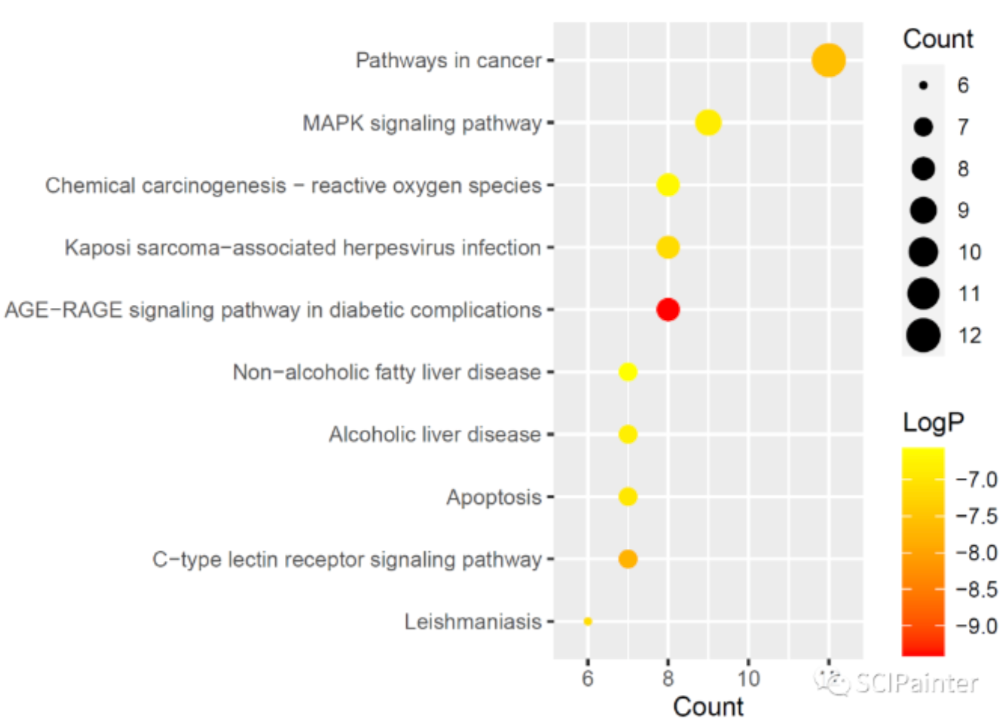
1 | #应用自带的主题 |
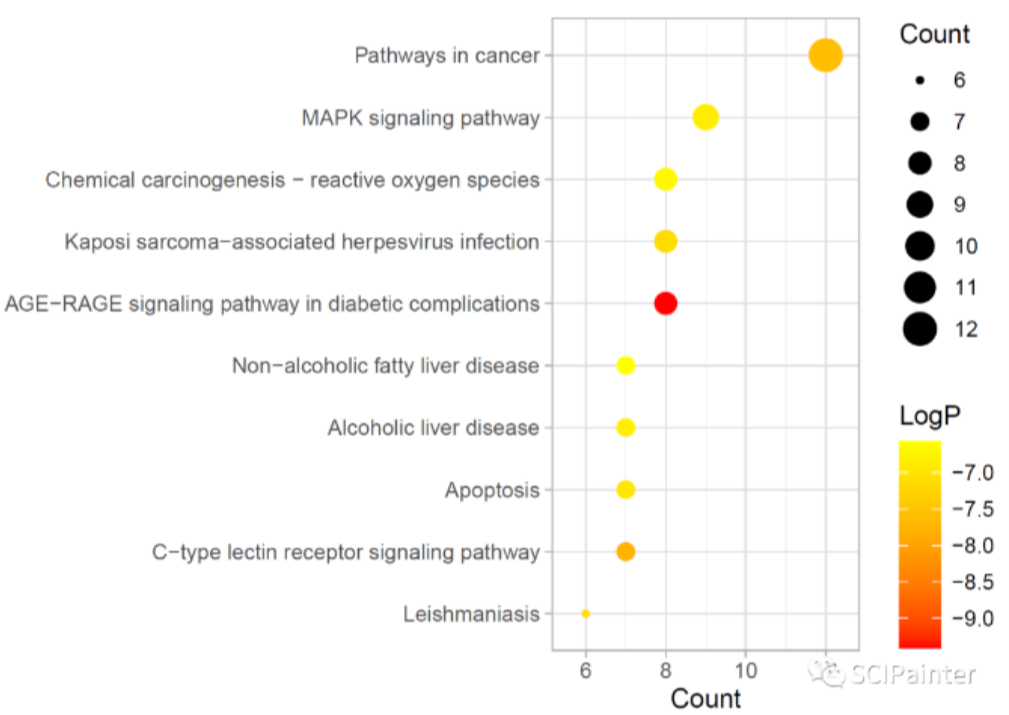
1 | #自定义图表主题,对图表做精细调整 |
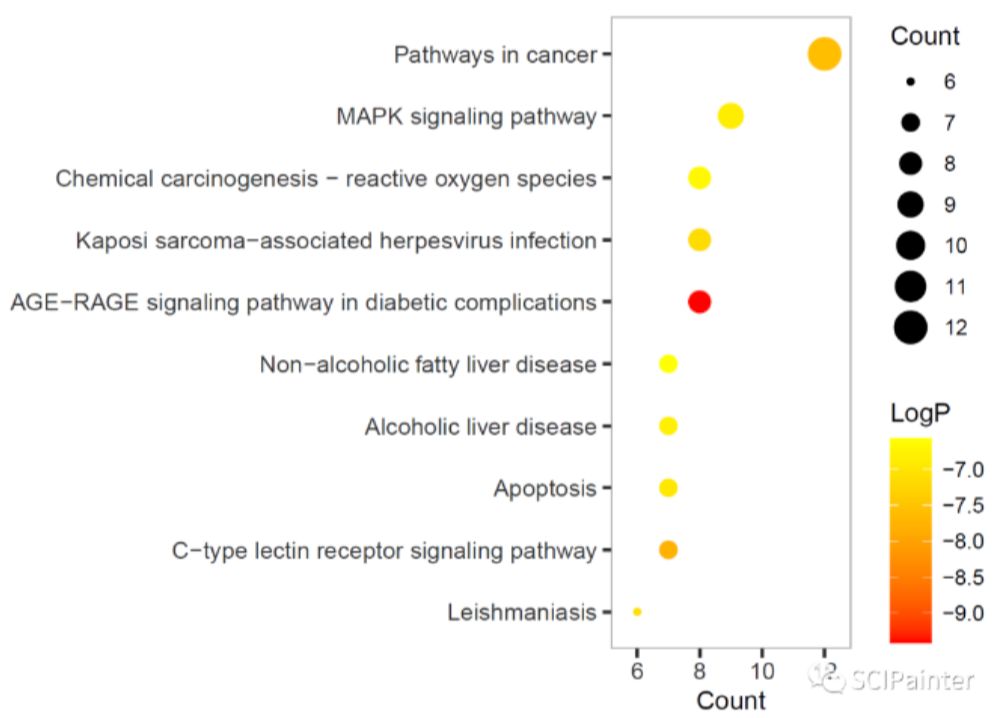
5.2 MetPA富集(Pathway topology analysis)
除了挖掘代谢途径内部的代谢物调控机制,对于整个生物体而言,代谢途径间的相互作用形成的大的调控网络也具备重要的研究意义。代谢途径间的相互调控是维持整个内部环境稳态的重要基础。 经过KEGG富集分析后,我们可以发现表型差异是由多个代谢途径共同作用导致的,这些代谢途径之间是否又有位置上或者说地位上的优先级?
MetPA富集分析,又叫做拓扑分析,以代谢途径未单位,但与KEGG富集不同,KEGG重点在解析目标代谢集主要参与的代谢途径是什么,筛选出显著富集的代谢物。
MetPA分析考虑生物体整体代谢网络,侧重突出在整个代谢网络中具备中药味点的代谢通路,结合统计分析检验核心代谢通路的显著性,再根据代谢途径内的信息,锁定重要的代谢物。
目前有在线的一些工具可以使用:MetaboAnalyst5.0
可以在线完成对人、小鼠、大鼠、奶牛、斑马、果蝇等15个物种的分析,使用目标代谢集上传分析(也就是之前提到的前景代谢集)
5.2.1 MetPA分析的原理
MetPA(Metabolic Pathway Analysis)是一种用于分析生物代谢网络中通路重要性的方法。在MetPA中,为了衡量或计算通路的位置信息,采用了两种中心性指标:Out-degree Centrality(出度中心性)和Relative-betweenness Centrality(相对中间中心性)。这些指标帮助研究者评估代谢通路在网络中的地位和作用。
Out-degree Centrality是评估网络中节点对外连通性的一个指标,它通过计算一个通路影响其他通路的数量来衡量其重要性。在代谢网络中,一个通路的Out-degree Centrality越高,表明它在网络中的影响范围越广,可能承担更为关键的功能。例如,在下图中,如果D通路能够影响B、C、E、F四个通路,那么D通路的Out-degree Centrality就是4,表示它具有较高的连通性和重要性。
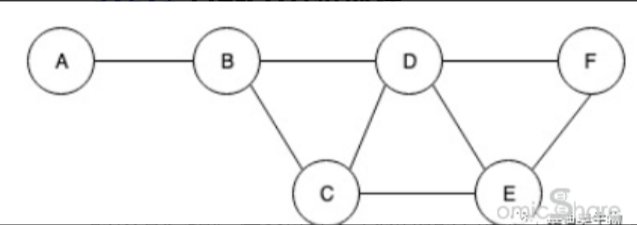
然而,Out-degree Centrality仅考虑了一个节点对其他节点的影响,而忽略了其他节点对该节点的影响。在代谢过程中,调控往往是双向的,即某些代谢物既可以作为反应的产物影响后续反应,也可以作为反应的底物被其他代谢物影响。因此,单独使用Out-degree Centrality可能无法全面评估一个通路在代谢网络中的真实地位。
为了更全面地评估通路的重要性,MetPA还引入了Relative-betweenness Centrality这一指标。Relative-betweenness Centrality衡量的是一个通路在所有其他通路对之间的最短路径上的出现频率。一个通路的Relative-betweenness Centrality越高,表明它在网络中的信息传递和控制作用越强,可能是代谢网络中的关键“桥梁”。简单理解就是,网络中当两个通路不能直接调控时,就需要借助第三个v通路才能建立调控关系。因此,如果整个网络中有更多的通路需要借助第三方通路v才能建立调控关系,v通路就崛北极高的重要性,但是v的连通性并不一定是最高的,只是相对位置是最重要的。他的计算方式如下:
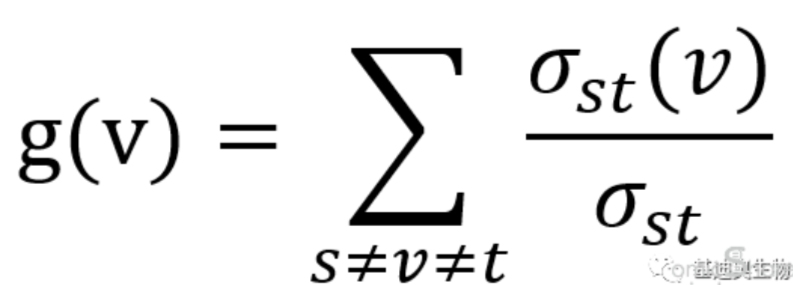
[^注]: g(v):指v通路的betweenness centrality; ost(v): 指s通路到t通路之间经过v的最短路径数; σst: 指s到t之间的所有最短路径数;
最后借助Fishers’Exact test、Hypergeometric Test、Globaltest等检验方式来判断通路的显著性,可以重点关注显著富集的通路。相比Out-degree Centrality,Relative-betweenness Centrality的统计方式更能从全局出发描述整个网络种通路相对位置的重要性。Relative-betweenness Centrality也是开发者非常推荐的统计方式。
MetPA富集分析实现方法: MetaboAnalyst5.0(https://www.metaboanalyst.ca/),无需注册,操作步骤如下:
登录网站,点击“Chick here to start“开始分析;
分析模块选择Pathway Analysis;

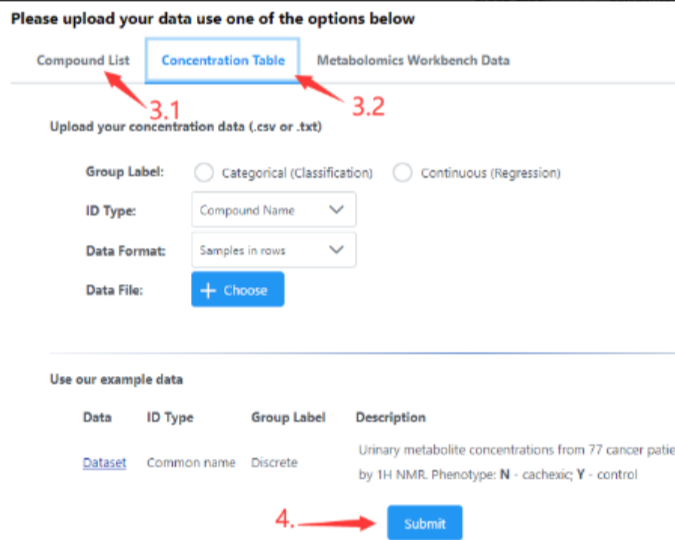
选择分析模式,可以只使用代谢物名称或者代谢物名称+丰度的矩阵数据进行分析。如果只关注代谢物,不关注丰度变化可选择使用代谢物名称(3.1)分析。若代谢物和丰度都关注,则选择3.2分析。该步骤会影响后续通路显著性的统计方式。
选择完成后点击“submit”
数据确认过程,可直接点击“Proceed”
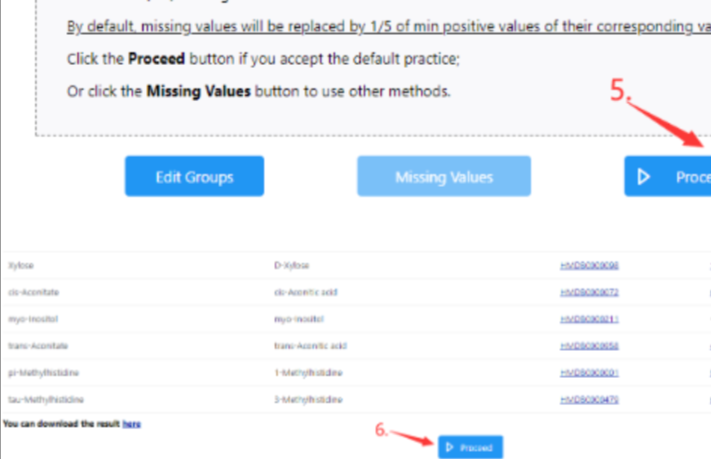
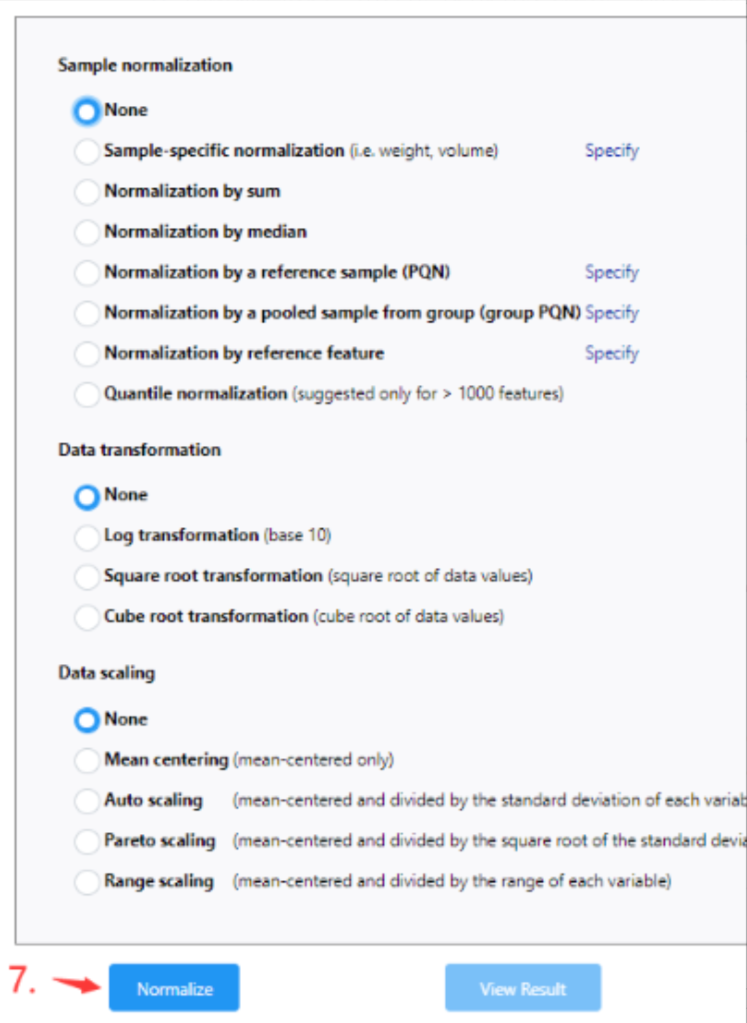
代谢物丰度归一化方式(若使用3.1模式,则没有此步骤),该步骤主要校正代谢物丰度数量级间的差异对富集结果的影响。可根据实际情况,选择是否需要归一化。(小Tips,实在纠结可以都试试,网页分析试错包容性强) “Normalize”后,点击“Proceed“后即可。
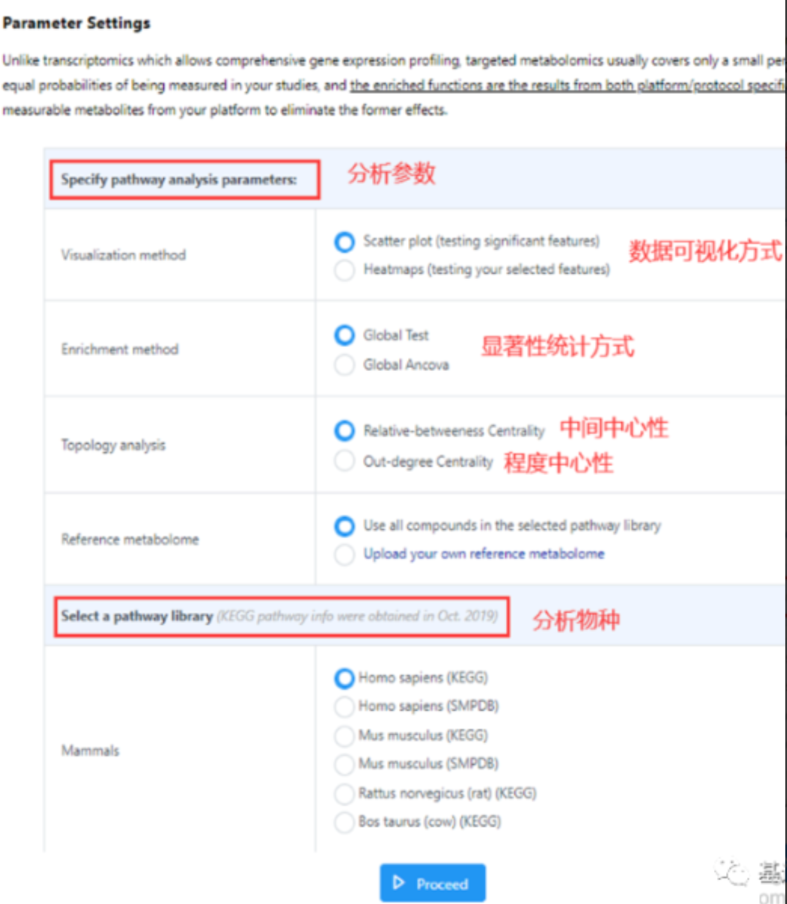
设置MetPA的富集方式。 这里使用默认参数(蓝色的圆圈)
- 获取分析结果。 点击左侧导航栏的“Download”,将结果下载到本地。 右侧未数据可视化区域, Pathway Impact是通路的位置信息,该值越大通路也就越重要,纵坐标为通路的显著性,颜色越红(越深)越显著富集。点击图中的圆点,右侧可出现通路的名称和富集到的代谢物信息。

5.3 MSEA富集分析(Metabolite Set Enrichment Analysis)
生物体内的代谢调控是层层递进的,极少会有一蹴而就的情况,之前提到的KEGG富集、MetPA富集,都是探讨目标代谢集主要腹肌道德代谢途径,这些分析方式往往存在一定的缺失,因为阈值过滤可能会损失一部分具备重要意义的微效代谢物的挖掘或发现。
例如下图中的pathway,可发现正是由于A的为上调,逐渐作用到下游代谢物,最终使E出现显著变化。 所以,该同类仍具备相当重要的生物学意义,如果根据差异倍数≥2,P<0.05来筛选差异代谢集,最终只能获得一个代谢物E,然后再结合KEGG富集、MetPA富集,最后得到的结果显然该pathway是不显著的。
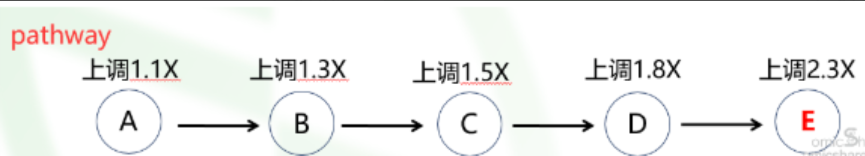
MSEA分析可弥补上述研究不足,重点挖掘代谢物丰度变化较低但仍对生物体调控具备主要意义的代谢物及其所在的代谢途径。其意义比较类似于转录组数据挖掘中的GSEA分析。
可使用MetaboAnalyst5.0网页(https://www.metaboanalyst.ca/)在线完成对人、小鼠、大鼠等物种的分析,分析时使用比较组中的所有代谢集开展,如A-vs-B得到的不经阈值筛选的所有代谢物。
5.3.1 MSEA分析的原理
MSEA再挖掘代谢途径时,有三种统计方式:Over Representation Analysis(ORA)、Single Sample Proling(SSP)、Quantitative Enrichment Analysis(QEA)模式。 ORA分析时,只需要输入比较组中的代谢物名称,无需包含丰度信息。 这种方式没有结合丰度数据,因此不利于丰度变化微弱的数据挖掘。
所以我跟推荐使用QEA模式开展分析。 QEA再分析通路时,借助广义线性模型计算每个代谢物浓度与样本的相关性,从而得到Q statistic,将pathway种每个代谢物Q statistic的平均值作为pathway的Q statistic,该值越高说明其与样本的相关性越强,对样本间的差异具有重要调控。 同时,软件还默认输出P值、Bonferroni校正后的P值和FDR值描述通路的显著性。
MSEA富集分析方法的实现:MetaboAnalysis5.0(https://www.metaboanalyst.ca/),操作步骤在网上都可查找到相关教程,这里不再赘述。
下期预告:蛋白代谢多组学研究思路
- 本文作者: Anderson
- 本文链接: http://nikolahuang.github.io/2024/04/05/代谢组学数据分析基本知识/
- 版权声明: 转载请注明出处,谢谢。