

[TOC]
- WES,WGS和WGRS的区别和联系是什么?
- WES (whole exome sequencing, 全外显子组测序): 是指利用序列捕获技术将【全基因组的外显子区域DNA】捕获富集后进行高通量测序,能够直接发现与蛋白质功能变异相关的遗传变异SNP(单核苷酸多态性)。以人类基因组为例,虽然外显子(蛋白质编码区)只占基因组的1%,但人类基因组85%的致病突变都在外显子区域,因此具有重要意义。
- WGS (whole genome sequencing, 全基因组测序): 是指对基因组整体进行高通量测序,分析不同个体间的差异,同时完成SNP及基因组结构注释。
- WGRS (whole genome re-sequencing, 全基因组重测序): 是指对已知参考基因组和注释的物种进行不同个体间的全基因组测序(WGS),并在此基础上对个体或群体进行差异性分析,鉴定出与某类表型相关的SNP.
通过比较可以发现,这三个测序技术其实都是在找基因组上的SNP(单核苷酸多态性), SNP分为四种:
- SNV(单核苷酸变异)
- InDel(插入缺失)
- SV(结构变异)
- CNV(拷贝数变异)
只是这三个技术的覆盖程度不同:
- WES:覆盖全基因组上的外显子区域
- WGS:覆盖全基因组
- WGRS:覆盖全基因组,是WGS在不同样本上的重复
只要你掌握了其中一种的测序分析与可视化,剩下的两种其实是一样的,本文就以WES测序分析与可视化为例(原因是手头只有RNA-Seq的转录组数据,勉强可以用来做WES数据的替代品吧),进行WES/WGS/WGRS的分析教学。
外显子是基因组中能够转录组出成熟RNA的部分。一个基因组中所有外显子的集合,即为外显子组。值得注意的是,通常所说的全外显子组测序,是指针对蛋白编码基因的外显子,很少涉及非编码基因。
基因(gene)是DNA中含有特定遗传信息的一段核苷酸序列的总称,是具有遗传效应的DNA分子片段,是控制生物性状的基本遗传单位。人类基因区间的大小可从数百个bp至超过200万个bp不等。根据人类基因组计划(The Human Genome Project)估计,人类拥有20000-25000个蛋白编码基因。
基因组(genome)指一个生物体所包含DNA的全部遗传信息。基因组由基因区域和非编码区域组成。**人类的基因组大小约为30亿个碱基对(bp)(3GB)**,其中非编码区域占到绝大多数,编码蛋白质的区域仅占约2%左右。
外显子组(exome)是基因组中所有外显子的集合。人类拥有约18万个外显子,约占人类基因组的1%,即约3000万个bp(30MB)。
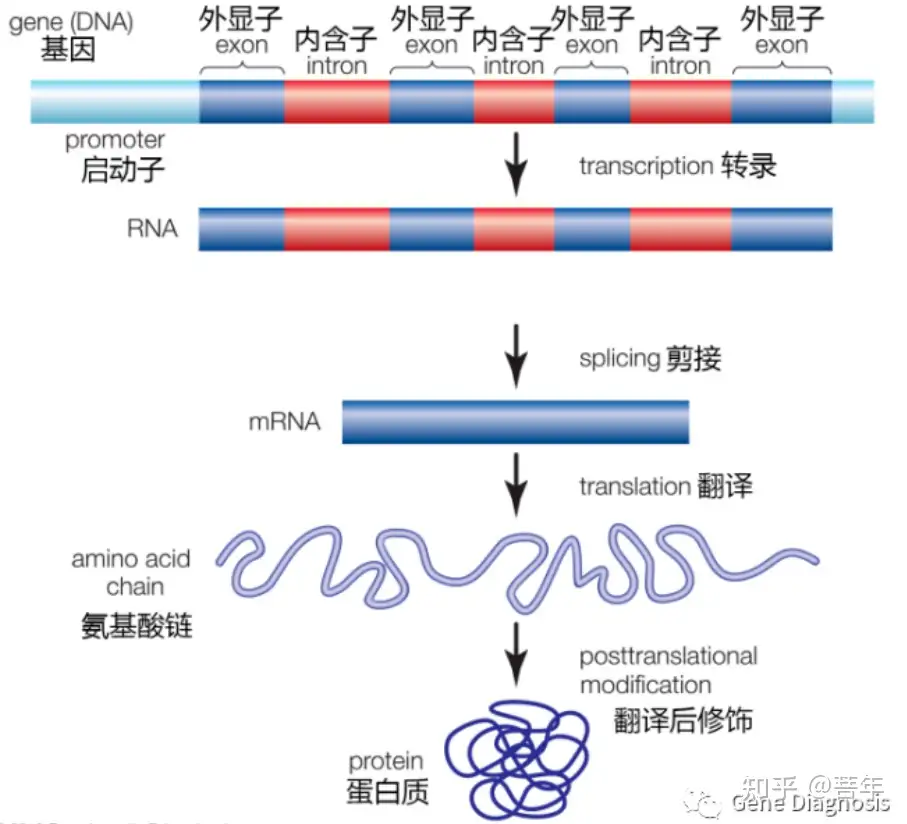
图:蛋白编码基因由内含子(非编码序列)和外显子(包括编码序列以及UTR区域)组成。要翻译有功能的蛋白,要进行以下步骤:基因从DNA转录为RNA前体,通过剪接形成成熟RNA,成熟RNA序列翻译成氨基酸链,以及蛋白质分子的翻译后修饰。
关于外显子,需要注意的一个特殊情况是非翻译区(UTR)。在mRNA的两侧分别存在5’UTR(前导序列)和3’UTR(尾部序列),它们的作用分别是调控翻译的启动和终止。它们由外显子序列构成,但不会被翻译成氨基酸。 所以,并非所有外显子序列都会被翻译成氨基酸。
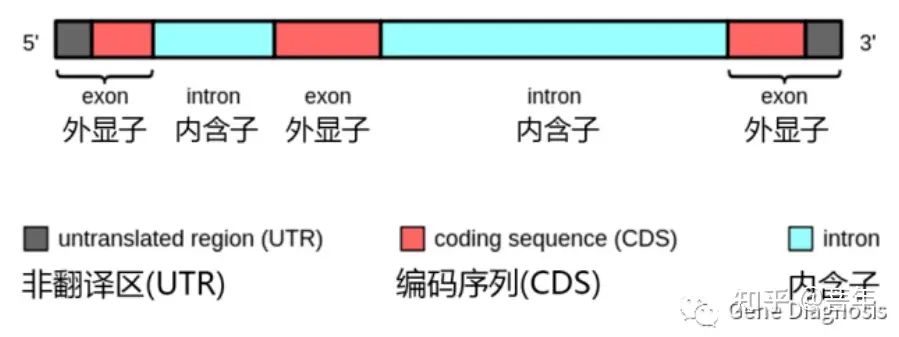
图:信使RNA前体(pre-mRNA)中的外显子。外显子既包括编码氨基酸的序列(红色),也包括不被翻译的序列(灰色)。
对外显子组(基因组里的所有外显子)进行测序的方法,即为 全外显子组测序 (Whole-Exome Sequencing,WES),也称为 外显子组测序、全外显子测序,全外测序 等。
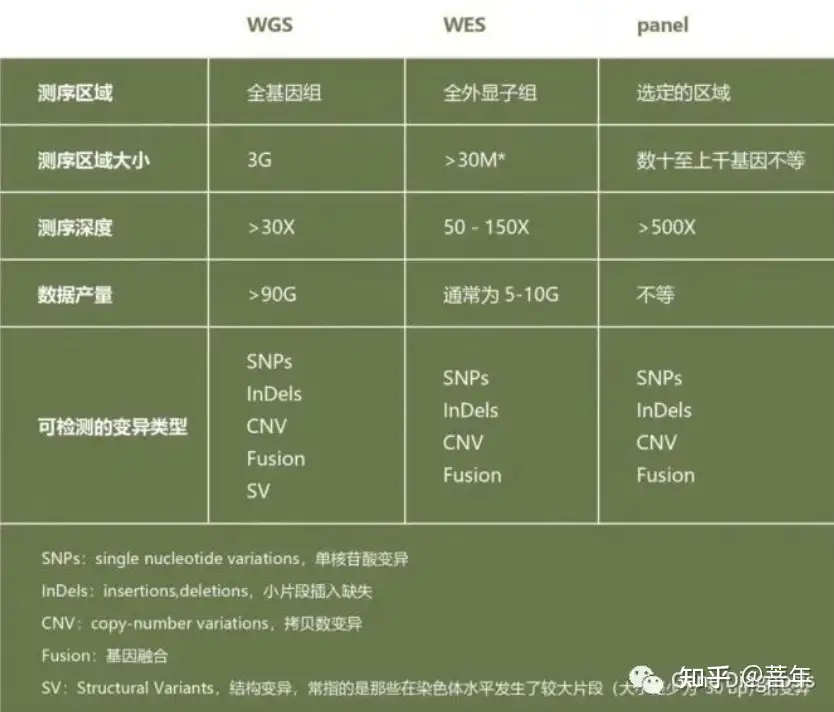
全基因组测序(Whole-Genome Sequencing, WGS)是对整个基因组进行测序。靶向测序(Targeted-sequencing,也称Panel sequencing)是对选定的基因进行测序,通常有几十个至一千个基因不等。因而,从覆盖基因组的范围来说,全基因组测序>全外显子组测序>靶向测序。
全外测序可以视作一种特殊的靶向测序——它靶向的区域是基因组上的所有外显子。
panel测序有两种技术原理:杂交捕获测序和多重扩增子测序。全外是基于序列杂交原理实现的
需要特别说明的是对CNV的检测。使用全外检测CNV时,在杂交捕获过程中,由于各个外显子的杂交效率不同,故不同外显子的覆盖率差异会较大。当出现阳性结果时,无法判断是由于杂交未捕获到,还是由于缺失。 故使用全外检测CNV容易出现假阳性结果。一般情况下,全外测序不用于CNV的检测 ,但在癌症研究中,利用癌组织和癌旁组织对照,可以检测体细胞CNV。
人类全外显子组所占基因组比例不超过2%,但它包含了约85%与疾病相关的变异,因此在研究编码基因变异层面,全外测序是比全基因组测序更为经济高效的替代方法。全外测序适用于孟德尔疾病、肿瘤、复杂疾病等多个研究领域。对于表现出异质性的疾病,或者患者表现出多个系统受累的复杂疾病症状时,尤为适合使用全外测序。
例如在肿瘤临床检测中,寻求肺癌靶向治疗的患者通常会先做panel测序,因为与肺癌靶向治疗相关的基因是比较明确的,几十至一百多个基因的panel测序通常就可以满足需求。而对于寻求免疫治疗的患者,通常会使用全外或大panel测序,来评估肿瘤突变负荷(Tumor Mutational Burden, TMB),TMB高的患者通常对免疫治疗有更好的响应。全外测序是业内公认的评估TMB的金标准。
*WES检测流程*
一个WES测序的工作流程,大体可以分为这3个部分:文库制备,测序,生信分析。
文库制备通常包含这些步骤:样本处理,DNA提取,定量,建库,杂交捕获,扩增,质控。
测序,目前的仪器包括国外Illumina公司测序平台,以及华大智造国产测序平台等。
生信分析的流程通常包含这些步骤:质控,拼接比对,去重和重排,变异检测,降噪和过滤,注释等。常用的软件有FastQC,BWA,GATK,ANNOVAR等。
一个完整的全外显子组测序,从样本处理到完成数据分析,通常需要10天左右时间。
捕获探针性能的评估指标
市面上的厂商会提供现成的全外探针,我们可以根据自己的需要来选择,也可以定制更为符合自己需要的“全外+”探针。
如果选择现成的探针产品,除了关注下面的探针评价指标外,也需要考虑探针大小、探针长度、探针设计等是否符合我们的样品和研究需求。
如果选择定制的产品,则可以在现成的探针产品上加入自己感兴趣的区域,也可以完全自主设计。在设计探针时常用的参考数据库有RefSeq、CCDS、Ensembl、GENCODE和ClinVar等。除了设计捕获区域外,还可以要求对某些特别的区域增加探针密度以提高捕获效率。
中靶率
中靶率(On-target rate)是一个百分数,用来表示测序数据中有多少能够比对到目标区域上。
在基因组上有许多与外显子有同源性的部分(比如内含子和基因间区),在实际工作中,这些并不属于目标(外显子)的部分在杂交过程中也会被捕获下来。这种探针捕获到非目标区域片段的情况称为脱靶(off target)。脱靶的数据是无效的,不能用于后续分析,即这部分测序数据被浪费了。
同等情况下,中靶率越高,由于脱靶产生的浪费越少,这款探针越好。
覆盖度
覆盖度(coverage)经常是和深度一起出现的,比如“10X覆盖度”、“30X覆盖度”。比如,“10X覆盖度为90%”指测序数据比对到目标区域后,有90%的区域被覆盖了至少10次,或者说有90%的区域有至少10条reads覆盖。
如果覆盖度没有和深度一起出现,则可以理解为“1X覆盖度”。比如“覆盖度为95%”,指95%的目标区域有至少1条reads覆盖到。换言之,有5%的目标区域连1条覆盖到的reads都没有,它们在这次测序中完全没被测到,被漏掉了。
同等情况下,覆盖度越高,越少比例的目标区域被漏掉,这款探针越好。
均一性
目标区域内不同的位点被覆盖的情况是不同的。比如一次WES测序的平均深度是60X,很有可能有的位点深度为10X,有的为40X,有的为90X这样的情况。均一性(uniformity)越好,即这些位点各自的深度越接近平均深度。
在实际工作中,我们根据期望达到的目标测序深度来分配数据量,即决定了这次测序的平均深度(平均深度=数据量/探针大小)。当某个区域的实际测序深度高于目标深度时,造成数据的浪费;而当某个区域的实际测序深度低于目标深度时,我们可能会认为这部分数据质量不好而丢弃它,导致这一区域无测序数据。均一性优良的探针可以帮助减少这两种情况的发生。
Fold-80是用来评价均一性的指标。它的定义是,为确保80%的目标碱基达到平均深度所需的额外测序的倍数。Fold-80越低,捕获效率越高,测序浪费越少。理想情况下的Fold-80为1。
Fold-80越低,均一性越好,越能节约测序成本,这款探针越好。
重复率
重复率(Dup rate)指的是重复序列(Duplicate reads)在总测序序列中的占比。由于这些重复序列不能带来额外信息,相反会影响变异检测结果准确性,因此需要在下游生信分析中去除这些重复序列。Dup rate越高,数据利用率越低,浪费的测序成本也就越多。
同等情况下,重复率越低,越能节省测序成本,这款探针越好
WES对于涉及拷贝数变异、非编码区变异和结构变异的疾病研究不适用。其次,在对目标区域进行捕获时, 存在捕获不均、捕获偏差等现象, 可以通过增加测序深度, 获得更多的序列信息进行统计分析, 以尽可能弥补这些偏差。
首先创建一个项目目录wes和存放每个步骤生成文件的子目录raw, clean, align, genome, hg19_VCF
1 | mkdir {raw,clean,align,genome,hg19_VCF} |
此流程包含两个raw外显子测序文件:wes.1.fq.gz,wes.2.fq.gz;存放于raw目录
原始数据质控,用fastp
1 | # 项目主目录下运行,在clean目录下生成两个过滤的fq文件 |
下载参考基因组(hg19)文件,存放于genome目录,并建立索引:
1 | for i in $(seq 1 22) X Y M; |
比对
1 | # clean目录下运行此命令,在align目录下生成sam文件 |
外显子区域覆盖度
需要生成外显子interval文件,生成这个文件的前提又需要dict文件和外显子bed文件(此处用的是安捷伦外显子bed文件,也可以去UCSC下载)。
1 | gatk CreateSequenceDictionary -R hg19.fa -O hg19.dict |
标记PCR重复序列并建立索引
以前用picard标记重复序列,现在这个工具全部整合到gatk中了
1 | nohup gatk --java-options "-Xmx10G -Djava.io.tmpdir=./" MarkDuplicates -I wes.sorted.bam -O wes.sorted.MarkDuplicates.bam -M wes.sorted.bam.metrics > log.mark 2>&1 & |
wes.sorted.bam.metrics有统计信息 wes.sorted.MarkDuplicates.bam创建索引文件,他的作用能够让我们可以随机访问这个文件中的任意位置,而且后面的步骤也要求这个bam文件一定要有索引。
变异检测
重新校正碱基质量值(BQSR)
变异检测是一个极度依赖测序碱基质量值,因为这个质量值是衡量我们测序出来的这个碱基到底有多正确的重要指标。它来自于测序图像数据的base calling,因此,基本上是由测序仪和测序系统来决定的,计算出来的碱基质量值未必与真实结果统一。 BQSR(Base Quality Score Recalibration)这个步骤主要是通过机器学习的方法构建测序碱基的错误率模型,然后对这些碱基的质量值进行相应的调整。 这里包含了两个步骤: 第一步,BaseRecalibrator,这里计算出了所有需要进行重校正的read和特征值,然后把这些信息输出为一份校准表文件(wes.recal_data.table) 第二步,ApplyBQSR,这一步利用第一步得到的校准表文件(wes.recal_data.table)重新调整原来BAM文件中的碱基质量值,并使用这个新的质量值重新输出一份新的BAM文件。
首先下载变异注释文件
1 | wget -c -r -nd -np -k -L -p ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg19 |
变异检测
HaplotypeCaller的应用有两种做法,差别只在于要不要在中间生成一个gVCF: (1)直接进行HaplotypeCaller,这适合于单样本,或者那种固定样本数量的情况,也就是只执行一次HaplotypeCaller。如果增加一个样本就得重新运行这个HaplotypeCaller,而这个时候算法需要重新去读取所有人的BAM文件,浪费大量时间; (2)每个样本先各自生成gVCF,然后再进行群体joint-genotype。gVCF全称是genome VCF,是每个样本用于变异检测的中间文件,格式类似于VCF,它把joint-genotype过程中所需的所有信息都记录在这里面,文件无论是大小还是数据量都远远小于原来的BAM文件。这样一旦新增加样本也不需要再重新去读取所有人的BAM文件了,只需为新样本生成一份gVCF,然后重新执行这个joint-genotype就行了。
推荐使用第二种,变异检测不是一个样本的事情,有越多的同类样本放在一起joint calling结果将会越准确,而如果样本足够多的话,在低测序深度的情况下也同样可以获得完整并且准确的结果。
1 | 第一种方法 |
变异质控与过滤
质控的含义和目的是指通过一定的标准,最大可能地剔除假阳性的结果,并尽可能地保留最多的正确数据。 第一种方法 GATK VQSR,它通过机器学习的方法利用多个不同的数据特征训练一个模型(高斯混合模型)对变异数据进行质控,使用VQSR需要具备以下两个条件: 第一,需要一个精心准备的已知变异集,它将作为训练质控模型的真集。比如,Hapmap、OMNI,1000G和dbsnp等这些国际性项目的数据,这些可以作为高质量的已知变异集。 第二,要求新检测的结果中有足够多的变异,不然VQSR在进行模型训练的时候会因为可用的变异位点数目不足而无法进行。
1 | gatk --java-options "-Xmx8G -Djava.io.tmpdir=./" VariantRecalibrator -R $GENOME -V wes.raw.vcf \ |
此方法要求新检测的结果中有足够多的变异,不然VQSR在进行模型训练的时候会因为可用的变异位点数目不足而无法进行。可能很多非人的物种在完成变异检测之后没法使用GATK VQSR的方法进行质控,一些小panel、外显子测序,由于最后的变异位点不够,也无法使用VQSR。全基因组分析或多个样本的全外显子组分析适合用此方法。
第二种方法通过过滤指标过滤。 QualByDepth(QD):QD是变异质量值(Quality)除以覆盖深度(Depth)得到的比值。 FisherStrand (FS):FS是一个通过Fisher检验的p-value转换而来的值,它要描述的是测序或者比对时对于只含有变异的read以及只含有参考序列碱基的read是否存在着明显的正负链特异性(Strand bias,或者说是差异性) StrandOddsRatio (SOR):对链特异(Strand bias)的一种描述. RMSMappingQuality (MQ):MQ这个值是所有比对至该位点上的read的比对质量值的均方根. MappingQualityRankSumTest (MQRankSum) ReadPosRankSumTest (ReadPosRankSum) 通过过滤指标过滤
1 | 使用SelectVariants,选出SNP |
突变注释
ANNOVAR是一个perl编写的命令行工具,能在安装了perl解释器的多种操作系统上执行。允许多种输入文件格式,包括最常被使用的VCF格式。输出文件也有多种格式,包括注释过的VCF文件、用tab或者逗号分隔的txt文件,ANNOVAR能快速注释遗传变异并预测其功能。这个软件需要edu邮箱注册才能下载。 ANNOVAR website
1 | 数据库下载 |
三种注释方式
**Gene-based Annotation(基于基因的注释)**:基于基因的注释(gene-based annotation)揭示variant与已知基因直接的关系以及对其产生的功能性影响。 Region-based Annotation(基于区域的注释):基于过滤的注释精确匹配查询变异与数据库中的记录:如果它们有相同的染色体,起始位置,结束位置,REF的等位基因和ALT的等位基因,才能认为匹配。基于区域的注释看起来更像一个区域的查询(这个区域也可以是一个单一的位点),在一个数据库中,它不在乎位置的精确匹配,它不在乎核苷酸的识别。基于区域的注释(region-based annotation)揭示variant与不同基因组特定段的关系。 Filter-based Annotation(基于过滤的注释):filter-based和region-based主要的区别是,filter-based针对mutation(核苷酸的变化)而region-based针对染色体上的位置。如在全基因组数据中的变异频率,可使用1000g2015aug、kaviar_20150923等数据库;在全外显组数据中的变异频率,可使用exac03、esp6500siv2等;在孤立的或者低代表人群中的变异频率,可使用ajews等数据库。 用table_annovar.pl进行注释,可一次性完成三种类型的注释。
1 | convert2annovar.pl # 将多种格式转为.avinput的程序 |
avinput文件由tab分割,最重要的地方为前5列,分别是: 1. 染色体(Chromosome) 2. 起始位置(Start) 3. 结束位置(End) 4. 参考等位基因(Reference Allele) 5. 替代等位基因(Alternative Allele) ANNOVAR主要也是利用前五列信息对数据库进行比对,注释变异。
SNP注释:
用table_annovar.pl进行注释,可一次性完成三种类型的注释。
1 | table_annovar.pl snp.avinput $humandb -buildver hg19 -out snpanno \ |
-buildver hg19 表示使用hg19版本 -out snpanno 表示输出文件的前缀为snpanno -remove 表示删除注释过程中的临时文件 -protocol 表示注释使用的数据库,用逗号隔开,且要注意顺序 -operation 表示对应顺序的数据库的类型(g代表gene-based、r代表region-based、f代表filter-based),用逗号隔开,注意顺序 -nastring . 表示用点号替代缺省的值 -csvout 表示最后输出.csv文件
Indel注释同上
测试数据:链接:https://pan.baidu.com/s/16tX5GrCLBAov9k_GjYYOyA 提取码:9pab
- 本文作者: Anderson
- 本文链接: http://nikolahuang.github.io/2024/03/17/WES数据分析流程-上/
- 版权声明: 转载请注明出处,谢谢。