

[TOC]
使用Maxquant对原始谱图搜库完成后,可以得到大量蛋白质的鉴定和定量信息。下一步就需要通过统计分析和生物信息学分析,从搜库得到的大量信息中筛选和寻找关键蛋白和信号通路,以及进行相关功能的验证。本讲我们会为大家介绍如何进行蛋白质组学的定量数据分析以及结果展示,主要包括:蛋白质鉴定和定量数据统计、差异表达蛋白筛选、功能注释和富集分析以及蛋白互作网络分析。
定量数据统计和展示
在蛋白质组学数据分析过程中,首先会对所有的鉴定和定量数据进行统计和展示,主要关注蛋白鉴定数目、肽段鉴定数目、肽段分布情况以及样品相关性和重复性等内容,展示蛋白质组学数据的定量深度、可信度和重复性等。
如下图所示,蛋白质组定量深度主要体现在各个样品中鉴定及定量到的蛋白和肽段数量,数量越多,表明定量深度越深,意味着能够获得更全面的蛋白质组学信息。同时,不同样品间定量深度的差异,也可以初步反映样品和方法的重复性和稳定性。
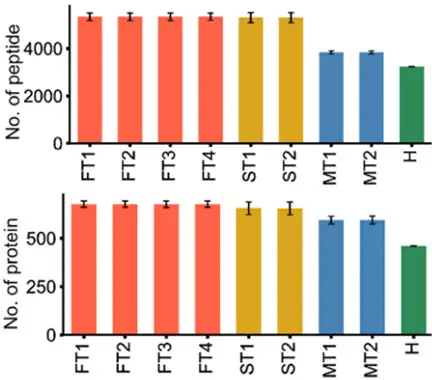
(肽段数量统计图(上)和蛋白数量统计图(下)[1])
蛋白质组学数据的可信度,可通过各个蛋白鉴定到的肽段数量以及各条肽段鉴定到的谱图数量分别展示。一般认为鉴定到2条及以上肽段的蛋白或2张及以上谱图的肽段为可信度更高的鉴定结果。
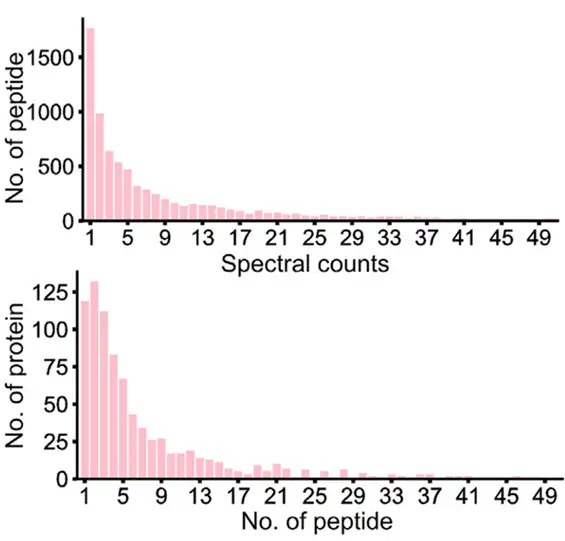
(谱图数量分布图(上)/肽段数量分布图(下) [1])
蛋白质组定量数据的相关性和重复性可通过相关系数来体现,组内样品间的蛋白质组定量相关系数往往较高,而组间样品的相关系数则会稍低(如左图)。PCA分析则是通过降维算法,将数百乃至上万个蛋白的定量数据降维为2个或3个反映定量特征的主成分,从而直观展示不同样品之间的定量相关性,在空间位置上接近的点表明其定量特征接近,也就是数据相关性更高。同一组的样品在PCA分析结果中往往会更为聚集(如右图)。
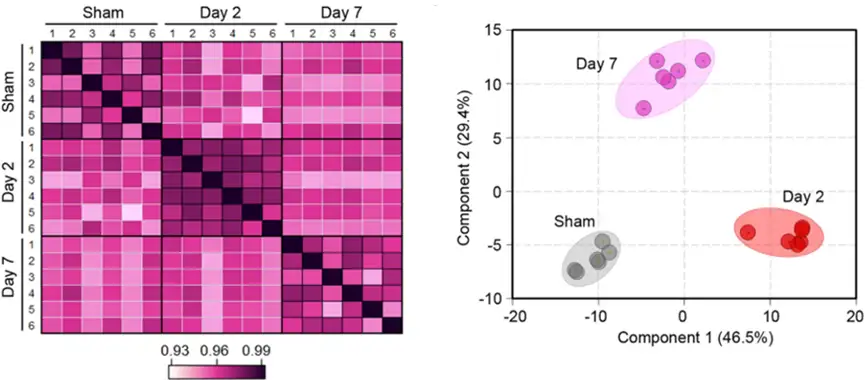
(相关系数热图(左)和PCA分析图(右)[2])
定量热图也是蛋白质组学中常用的展示定量数据的结果图形。在定量热图中,通常每一列代表一个样品,每一行代表一个蛋白,以色块颜色表示各个样品中蛋白的定量水平。通过颜色的变化和分布可以直观的展示不同样品中蛋白定量水平的相关性和差异性。
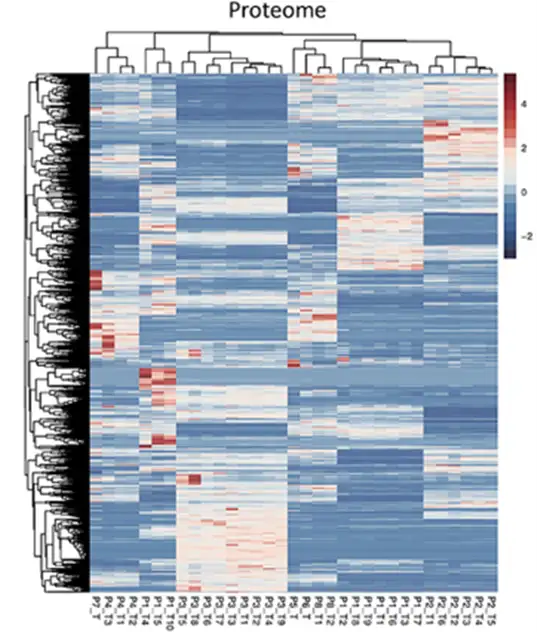
(蛋白定量热图[3])
差异表达分析
找出不同处理组间差异表达蛋白是蛋白质组学研究的重要目标之一。统计学上一般选取P值(P-value)和差异倍数(Fold change)两个参数实现差异蛋白的筛选,其中P-value由比较两组样品蛋白定量数据的T检验计算得出,要求各组样品数量不少于3个;Fold change是两样品中同一个蛋白的表达差异倍数,主要根据蛋白定量值计算。如下图所示,根据显著性P<0.05和 Fold Change>1.5或<1/1.5设定阈值可挑选出符合标准的差异蛋白质。
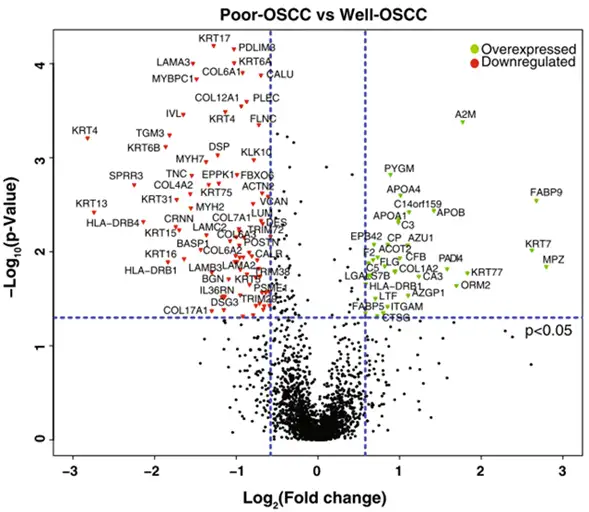
(差异蛋白火山图[4])
筛选出差异表达蛋白后,也可再次通过定量热图与PCA分析图,更为直观地展示这些差异蛋白的定量特征,以及各样本组内及组间的相似和变异程度。
功能注释和富集分析
以上分析完成后,有些实验还需要对蛋白的功能、分布、参与的通路等方面进行注释和富集,可为后续生物学和功能机制的研究提供参考和线索。
蛋白功能注释是将生物学信息关联到蛋白质序列的过程,使用的数据库主要有GO(gene ontology)、KEGG(Kyoto encyclopedia of genes and genomes)、COG(Clusters of Orthologous Groups of proteins)、HallmarkGeneset、Reactome等。以GO分析为例,可理解为将蛋白对应的基因分门别类地放入一个个功能条目的盒子中去,从功能(Molecular Function)、参与的生物途径(Biological Process)和细胞中的定位(Cellular Component)三个维度来较为全面地描述一个蛋白。如下图GO注释,饼图的每一个分区表示一个GO条目,分区大小表示本条目中蛋白数量在差异蛋白中的占比。
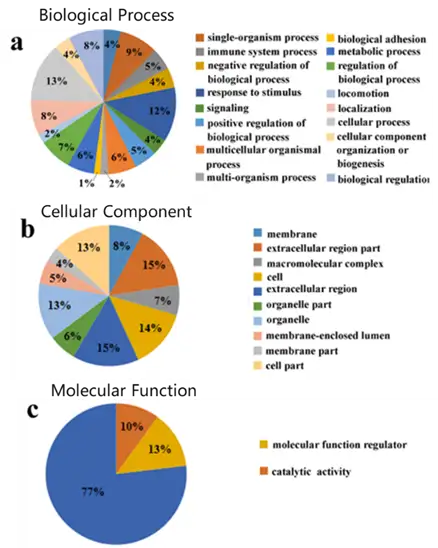
(GO注释饼状图[5] )
富集分析则是在功能注释的基础上的进一步分析,其主要目的是以背景蛋白为参照,分析差异蛋白较为集中分布的注释条目,从而筛选出与差异蛋白更相关、更可能具有生物学意义的功能注释条目。目前最常用的富集分析算法是基于超几何分布的Fisher精确检验。
富集分析通常以P<0.05作为条目显著富集的筛选阈值。富集得到的注释条目可通过如下柱状图进行展示。其中GO富集分析通常会以不同颜色表示GO条目所属大类。
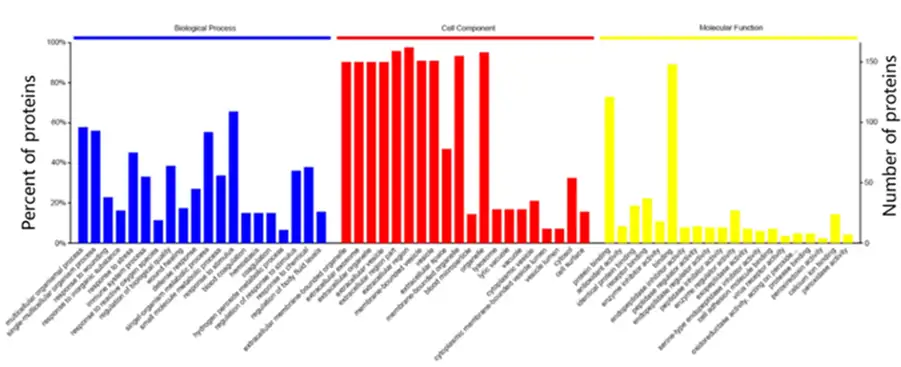
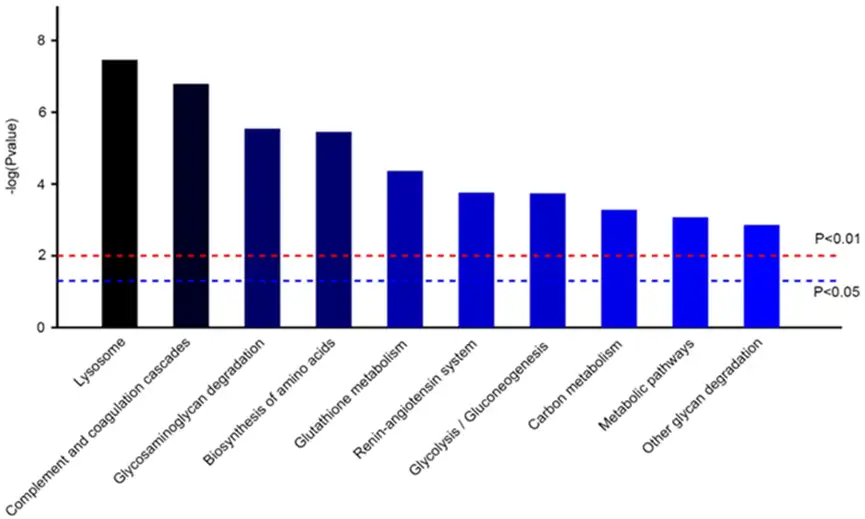
(GO富集柱状图(上)和KEGG富集柱状图(下)[6])
气泡图也是富集分析常见图形展示,相比柱状图可以通过气泡的横坐标、颜色、大小展示更多维度的富集参数,包括E-ratio(富集倍数)、p值和条目中蛋白数量等。其中富集倍数越大、p值越小说明该条目在差异蛋白中富集程度越显著。
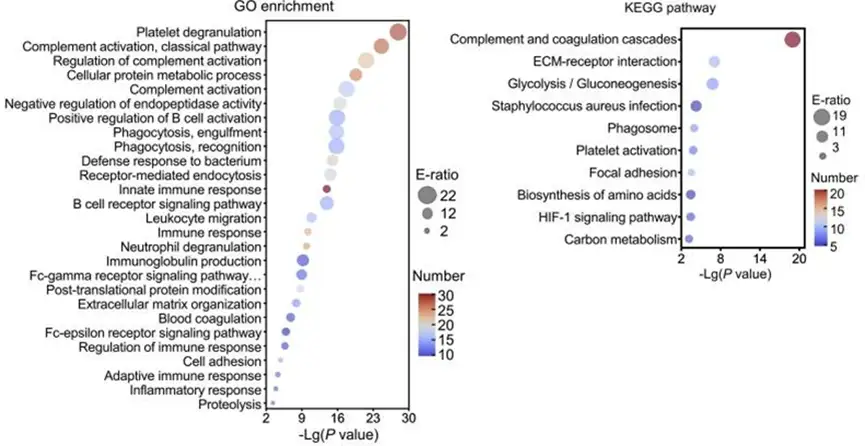
(GO富集气泡图(左)和KEGG富集气泡图(右)[1])
互作网络分析
在生物体内,蛋白质并非单独发挥作用,其功能的行使须依赖于其他蛋白的调控和介导,这种调控和介导要求蛋白质之间存在直接或间接的相互作用。
STRING(Search Tool for the Retrieval of Interaction Gene/Proteins)数据库是目前使用最广泛、信息最全面的蛋白互作网络分析工具,既包含了已有报道的蛋白质互作数据,也包含了基于预测的蛋白质互作信息。文章中一般以PPI(Protein‑Protein Interaction)互作网络图进行展示,每个节点为一个差异蛋白,节点之间的连线表示两个蛋白之间存在已知的或预测可能存在的蛋白相互作用,节点颜色表示该蛋白在比较组中的定量信息,如下图所示。PPI互作网络可以直观的展示蛋白间的相互关联,并进一步帮助我们寻找其中的关键节点。
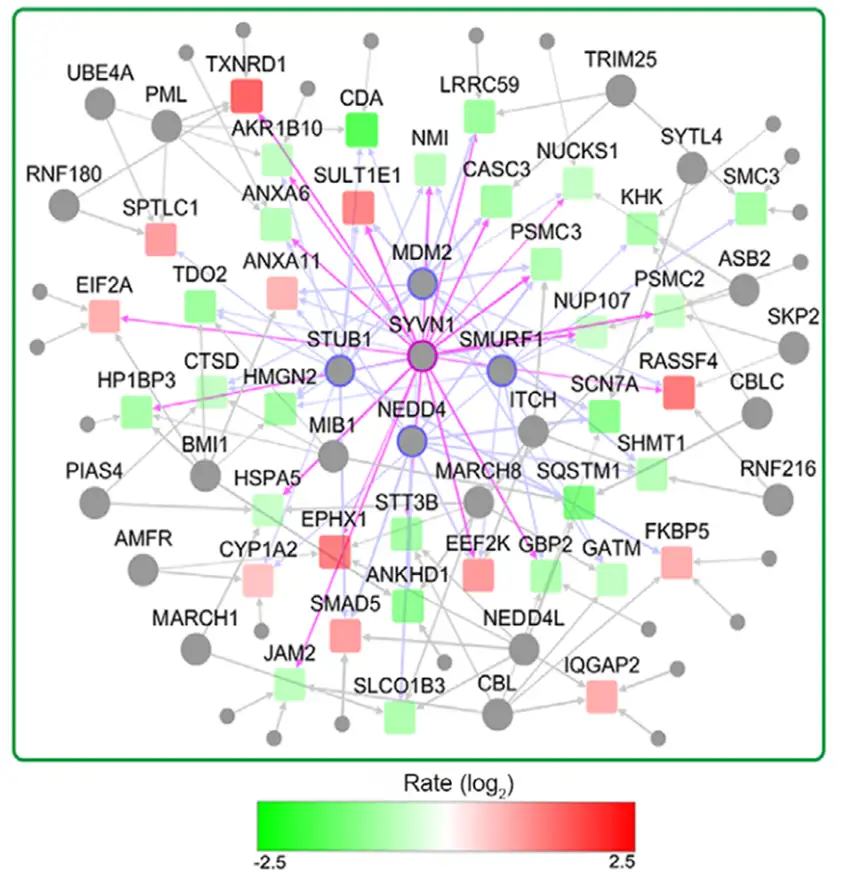
(蛋白互作网络图[7])
总结
蛋白质组学的数据分析通常包含以下内容:通过比较不同蛋白质在不同样本中的表达情况,找到差异表达蛋白;通过功能注释和富集分析,找到差异蛋白显著富集的生物学功能和信号通路;对差异表达蛋白进行蛋白互作网络分析,展示差异蛋白间的相互关联,从而寻找网络中的关键节点。这些分析能够帮助我们筛选获得关键蛋白、生物学功能和信号通路,对于深度解析蛋白质组学数据,指导后续功能研究等具有重要意义。
参考文献
- Shu T, Ning W, Wu D, et al. Plasma Proteomics Identify Biomarkers and Pathogenesis of COVID-19[J]. Immunity, 2020, 53(5): 1108-1122
- Lin Y H, Platt M P, Fu H, et al. Global Proteome and Phosphoproteome Characterization of Sepsis-induced Kidney Injury[J]. Mol Cell Proteomics, 2020, 19(12): 2030-2047.
- Zhang Q, Lou Y, Yang J, et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas[J]. Gut, 2019, 68(11): 2019-2031.
- Mohanty V, Subbannayya Y, Patil S, et al. Molecular alterations in oral cancer using high-throughput proteomic analysis of formalin-fixed paraffin-embedded tissue[J]. Journal of cell communication and signaling, 2021, 15(3): 447-459.
- Wen L, Liu Y F, Jiang C, et al. Comparative Proteomic Profiling and Biomarker Identification of Traditional Chinese Medicine-Based HIV/AIDS Syndromes[J]. Sci Rep, 2018, 8(1): 4187-4197.
- Zhou F, Luo Q, Han L, et al. Proteomics reveals urine apolipoprotein A-I as a potential biomarker of acute kidney injury following percutaneous coronary intervention in elderly patients[J]. Exp Ther Med, 2021, 22(1): 745-755.
- Ji F, Zhou M, Sun Z, et al. Integrative proteomics reveals the role of E3 ubiquitin ligase SYVN1 in hepatocellular carcinoma metastasis[J]. Cancer Commun (Lond), 2021, 41(10): 1007-1023.
- 本文作者: Anderson
- 本文链接: http://nikolahuang.github.io/2024/03/17/蛋白质组学数据分析和展示/
- 版权声明: 转载请注明出处,谢谢。