

[TOC]
HZ Hanwen Zhang,SZ Siwei Zhang ![]()
第一次发布: 2021年06月20日第11卷第12期 DOI: 10.21769/BioProtoc.4051
评审: Jaira Ferreira de VasconcellosGiovanna Piovani 匿名评审
Abstract
人类诱导多能干细胞(hiPSCs)在发育生物学和疾病建模领域被广泛使用。在iPSC线中使用CRISPR/Cas9基因编辑通常具有低频率,这阻碍了其在疾病相关单核苷酸多态性(SNPs)的精确等位基因编辑中的应用,尤其是在基因组非编码部分。在这里,我们介绍了一种独特的流程,通过SNP编辑将异源二倍体iPSC线转化为疾病风险等位基因或非风险等位基因的同源二倍体,使用一种临时且简单的基于转染的协议。此协议使我们能够在约4至5周内同时获得SNP位点所有三种可能基因型的纯合和克隆同源系。
背景介绍
将CRISPR/Cas9基因编辑应用于iPSC细胞,在发育生物学、疾病建模和再生医学等领域提供了无与伦比的前景。然而,由于iPSCs对传统CRISPR/Cas9基因编辑策略的适应性不强,编辑效率通常较低,尤其是对于通过同源定向修复(HDR)介导的单核苷酸多态性(SNPs)编辑。必须开发特殊的实验方法来克服这一限制;例如,同时使用CRISPR/Cas9突变和iPSCs的重编程(Howden等,2015;Tidball等,2018),先进的细胞分选技术如FACS(Forsyth等,2006;Miyaoka等,2014),或通过靶向质粒中长同源臂的存在重组携带突变的载体(Hendel等,2014)。尽管这些高度专业化的iPSC线基因编辑协议在时间上和/或资源上都是消耗性的,这仍然是通过CRISPR/Cas9介导的精确SNP编辑生成同源iPSC线的一个关键限制因素。
在这里,我们提出了一种经过验证的工作流程和详细程序,允许直接使用基于脂质体的CRISPR/Cas9系统进行HDR编辑,通过抗生素选择富集编辑的iPSC,并通过单克隆选择获得所有三种可能基因型的纯合iPSC同源线。该方法改编自Ran等(2013)的工作,并在我们最近的出版物中得到了证明(Forrest等,2017;Zhang等,2018和2020)。值得注意的是,该协议对于从异源二倍体iPSC线到疾病风险等位基因或非风险等位基因的同源二倍体的独特CRISPR/cas9 SNP编辑设计非常有效,这使得通过直接比较所有三种不同基因型的同源线,对编辑的SNP的功能解释更加可靠(Forrest等,2017;Zhang等,2018和2020)。
材料和试剂
A. 材料
- 1.5 ml Eppendorf管(VWR,货号:89000-028)
- 4-w NunclonTM Delta多孔板(Thermo Scientific,货号:62407-068)
- 6孔培养板(Thermo Fisher,货号:140675)
- 96孔培养板(Corning,Falcon®,货号:353072)
- 标准60 × 15 mm带通风孔的培养皿(Fisher Scientific,货号:12-565-95)
- 15 ml离心管(VWR,货号:21008-216)
- 2 ml带盖的冷冻管,聚丙烯(Corning®,货号:89089-764)
- 10 μl排管,低保留率无菌(VWR,货号:10017-062)
- 200 μl排管,低保留率无菌(VWR,货号:76322-150)
- 1,000 μl低保留率排管(VWR,货号:10017-090)
- BioDot 96孔非裙边PCR板(Dot Scientific,货号:650-PCR)
B. 试剂列表:
- mTeSR1(STEMCELL,目录号:85850)
- mFreSR 冷冻保存培养基(STEMCELL,目录号:05855)
- ReLeSR(STEMCELL,目录号:05872)
- Matrigel® hESC-Qualified Matrix(Corning®,目录号:354277)
- FuGENE® HD 转染试剂(Promega,目录号:E2311)
- Accutase(STEMCELL,目录号:07920)
- Primocin(Invitrogen,ant-pm-1)
- Y-27632 二氢氯酸盐(R&D Systems,目录号:1254/1)
- QIAprep Spin Miniprep Kit(250)(Qiagen,目录号:27106)
- Qiaprep EndoFree Plasmid Maxi Kit(10)(Qiagen,目录号:12362)
- BigDye Terminator v3.1 循环测序试剂盒(Thermo Fisher Scientific,目录号:4337455)
- DyeEx 2.0 试剂盒(Qiagen,目录号:63204)
- HotStarTaq DNA聚合酶(250 U)(Qiagen,目录号:203203)
- PlasmidSafe Exonuclease(Lucigen,目录号:E3101K)
- QuickExtractTM DNA提取溶液(VWR,目录号:76081-768)
- FastDigest BbsI(Thermo Fisher Scientific,目录号:ER1101)
- T7连接酶(NEB,目录号:M0318S)
- ATP溶液,100 mM(NEB,目录号:N0451)
- dNTP溶液混合物,10 mM(NEB,目录号:N0447S)
- Opti-MEM I,100 ml(Thermo Fisher Scientific,目录号:31985062)
- 虾碱性磷酸酶(Thermo Fisher Scientific,目录号:783901000UN)
- PCRX Enhancer System(Thermo Fisher,目录号:11495017)
- 异丙醇(Sigma-Aldrich,目录号:190764)
- 核酸酶-free水,PCR级(Thermo Fisher Scientific,目录号:AM9937)
- Teknova DNA/RNA Resuspension Buffer(TE缓冲液)(VWR,目录号:100216-886)
- Invitrogen One Shot® Stbl3TM 化学感受态大肠杆菌细胞(Thermo Fisher Scientific,目录号:C737303)
- 1 kb DNA梯形标志物(Promega,目录号:G5711)
设备列表:
- C1000 Touch 热循环仪(Bio-Rad,型号:1851197)
- Sorvall Legend XTR 离心机(Thermo Scientific,目录号:75217420)
- 台式冷冻离心机 5430R(Eppendorf,目录号:022620601)
- Heracell 150i 组织培养孵育箱(Thermo Fisher Scientific,目录号:51026283)
- ABI 3730 DNA分析仪(Thermo Fisher Scientific,型号:3730S)
- Nalgene Mr. Frosty 冷冻容器(Sigma-Aldrich,目录号:C1562)
软件: - ApE – A plasmid Editor(M Wayne Davis,https://jorgensen.biology.utah.edu/wayned/ape/)
实验步骤
A. 实验规划
细胞系选择
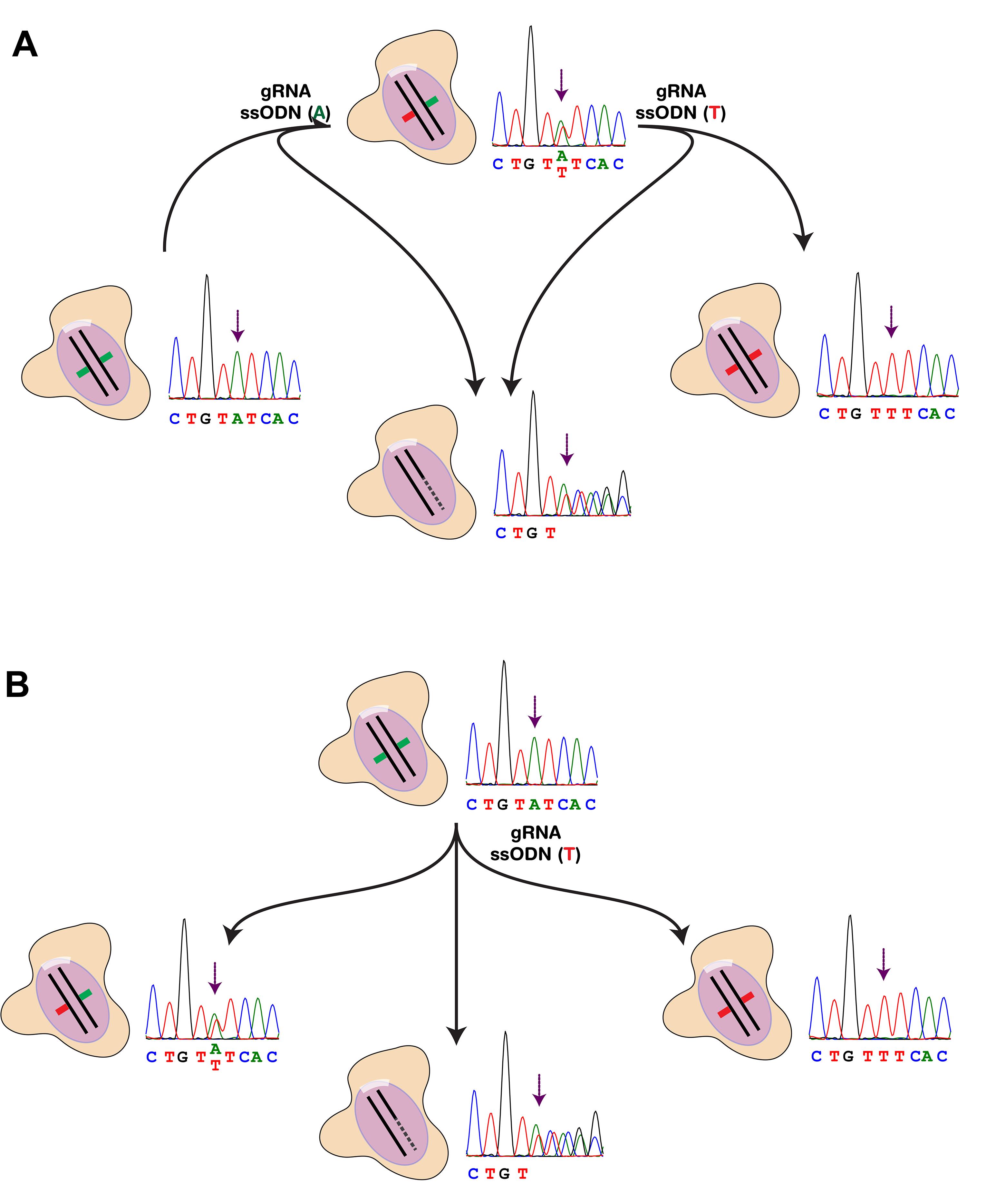
通常情况下,如果起始细胞线在要编辑的指定SNP位点为杂合子,实验将更为方便。这样,我们可以合成两种单链DNA寡核苷酸(ssODN),每种携带不同的基因型,并同时进行两个独立的SNP编辑实验,最终在实验结束时获得两个方向上均为纯合子的编辑细胞系(图1A)。在条件不允许高度自由选择时,例如使用来自临床诊断患者的细胞系,从纯合子细胞系开始也是可能的(图1B)。在这种情况下,增加在E和F部分挑选的克隆数量将有助于最大化发现阳性克隆的机会。尽管在第一轮CRISPR/Cas9编辑中,杂合子或相反基因型的纯合子可能不会出现,但通常获得相反基因型的纯合子细胞系对于实验目的来说已经足够满意。或者,可以使用从第一轮编辑中获得杂合子细胞系,再用相同的ssODN进行一轮CRISPR/Cas9编辑,以获得相反基因型的纯合子。在所有情况下,在继续进行B部分之前,需要确认起始细胞线在所需SNP位点的基因型。为了确认特定SNP位点的基因型,使用从起始细胞系提取的30 ng基因组DNA,并按照E4-E5步骤进行Sanger测序。另外,在线数据库、下一代测序结果或与细胞系相关的患者元数据也可能提供要编辑的SNP位点的基因型信息。
技术要求
整个实验包括三个主要部分:质粒克隆与准备;诱导多能干细胞(iPSC)的维护、转染和亚克隆;以及Sanger测序。虽然理想的情况是操作者熟悉所有这些技能,但本协议已被设计为几个成员的合作项目,每个成员都有自己的专长。
B. gRNA设计与载体准备
使用dbSNP(https://www.ncbi.nlm.nih.gov/snp/)或任何基因组浏览器获取目标SNP附近的DNA序列以进行gRNA设计。通常,序列将延伸目标SNP上下游各65个碱基,总共131个碱基。保存此序列,因为它将用于合成带有目标SNP位点两侧同源臂的ssODN。此序列还将在Benchling(https://www.benchling.com)等gRNA设计工具中使用,生成用于引入双链断裂(DSB)的gRNA序列(20个碱基)。选择效率评分最高的gRNA,同时保持合理的特异性评分。还发现,如果gRNA序列的切割位点靠近目标SNP位置,可能会获得更好的重组效率(见图2,展示了用于编辑SNP rs2027349的gRNA和ssODN的Benchling示意图和输出示例)。同时,使用Primer3(http://primer3.ut.ee/)或类似在线工具设计一对覆盖目标SNP的PCR引物。PCR产物的大小应为400-600个碱基,且SNP至少距离产物任一端150个碱基。
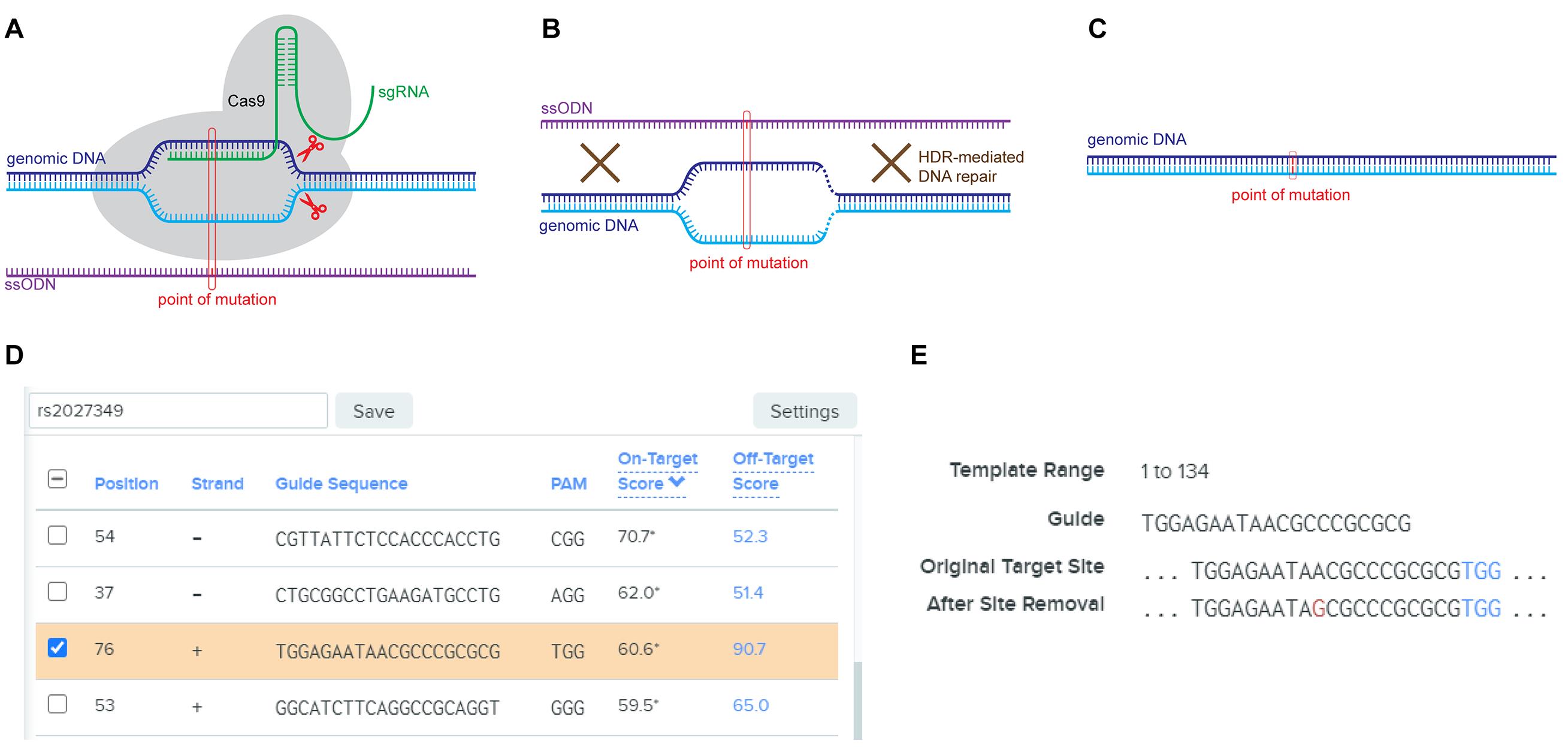
[^图2展示了通过HDR(同源定向修复)介导的SNP编辑的示意图。]: A部分显示了gRNA引入的双链断裂事件。单向导RNA(sgRNA)与特定的基因组DNA序列结合,并在基因组DNA双链中引入双链断裂。图中平行显示了ssODN,以突出突变位点。B部分展示了使用ssODN作为模板的HDR介导的DNA修复事件。在创建双链断裂(虚线)后,HDR介导的DNA修复开始,在此过程中,某些细胞使用携带突变等位基因的ssODN作为最终修复产物的模板。C部分显示了使用ssODN作为模板修复后的最终产品,突出了突变点。D部分是从Benchling输出的样本gRNA候选列表,使用的是人类GRCh38基因组序列chr1:150,067,554-150,067,687。这里突出显示的项目包含SNP rs2027349,其序列为…AATA[A]CGCC…。E部分显示了从Benchling输出的样本ssODN窗口,使用的是上述相同的基因组序列,展示了SNP本身从A到G的转换(红色突出显示)。
- ssODN,长度为131个核苷酸,单链,编辑等位基因的目标SNP位于碱基位置66。不要订购互补链。如果从杂合子细胞系开始,需要两种携带两种等位基因的ssODN;如果从纯合子细胞系开始,则需要一种携带相反等位基因的ssODN。如果从IDT订购,请订购4 nmol Ultramer Oligo干粉格式,标准脱盐。
- sgRNA-top:5’-[Phos]-CACCgNNNNNNNNNNNNNNNNNNNN;
- sgRNA-bottom:5’-[Phos]-AAACNNNNNNNNNNNNNNNNNNNNc;
其中N代表gRNA序列。注意sgRNA-bottom寡核苷酸末尾的c。sgRNA寡核苷酸中不要包含PAM序列。 - 两个寡核苷酸都可以订购5′-磷酸化格式(推荐),以避免内部PNK磷酸化步骤(其效率不一)。如果从IDT订购,请订购25 nmol干DNA Oligo格式,标准脱盐,并在到达后用TE缓冲液重新溶解至100 μM,长期储存于-20°C。
- 如上所述的测序引物。如果SNP离任一端不太近/远,PCR引物可以用作测序引物。订购25 nmol干DNA Oligo格式,标准脱盐,并在到达后用TE缓冲液重新溶解至100 μM,长期储存于-20°C。
- U6-Forward引物(5’-gagggcctatttcccatgatt)用于测序最终质粒构建中的gRNA插入物。订购25 nmol干DNA Oligo格式,标准脱盐,并在到达后用TE缓冲液重新溶解至100 μM,长期储存于-20°C。
组装pSpCas9(BB)-2A-Puro (PX459) V2.0(Addgene,62988)构建体与定制gRNA插入物。
a. 在室温下,将以下成分在带有盖子的PCR管中彻底混合。在整个实验过程中不要使用二乙基吡咯烷酮(DEPC)处理的水,因为它可能会抑制反应。
组件 量(μl)
sgRNA top (100 μM) 1
sgRNA bottom (100 μM) 1
T4连接缓冲液,10×(NEB) 1
水,PCR级 7
总计 10b. 使用带有加热盖的热循环器,并设置以下参数以退火寡核苷酸。以最小升温速率(例如,0.1°C/s)降温。
步骤# 温度 时间 备注
1 95°C 5分钟 最小升温速率
2 25°C 2分钟c. 稀释退火的寡核苷酸,方法是将1 μl加入到199 μl PCR级水中,彻底混合。
d. 在PCR管中混合以下成分以进行切割连接反应。我们建议通过用水替换稀释的寡核苷酸来设置一个阴性对照实验,其功能将在后面介绍。
组件 量(μl)
pSpCas9(BB)-2A-Puro (PX459) V2.0, 100 ng/μl 1
步骤A3c中稀释的退火寡核苷酸 2
Tango缓冲液,10× 2
DTT,10 mM 1
ATP,10 mM 1
FastDigest BbsI 1
T7连接酶 0.5
水,PCR级 11.5
总计 20然后设置热循环器的参数如下:
步骤# 温度 时间 备注
1 37°C 5分钟
2 21°C 5分钟
3 / / 返回到步骤#1,6个循环e. 将连接产物和阴性对照与PlasmidSafe外切酶混合,以消化任何残留的线性化DNA。将以下成分在带有盖子的PCR管中组装并彻底混合。这个热灭活的反应如果无法立即使用,可以在-20°C下储存至少1周或在-80°C下无限期储存。
组件 量(μl)
步骤A3d中的连接产物 11
PlasmidSafe缓冲液,10× 1.5
ATP,10 mM 1.5
PlasmidSafe外切酶 1
总计 15然后设置热循环器如下:
步骤# 温度 时间 备注
1 37°C 30分钟
2 70°C 30分钟
3 4°C 保持f. 转化感受态细胞。我们建议使用RecA-的大肠杆菌菌株,如Invitrogen Stbl3(recA13)或NEB Stable(recA1),因为插入的gRNA及其相关的gRNA支架可能包含不稳定序列。作为指导,按照供应商的说明,每转化使用步骤A3e中的2 μl产物(包括阴性对照)。热冲击后,需要加入复苏培养基(如SOC或NEB 10-beta/Stable Outgrowth Medium),在32°C下摇床培养1小时,然后铺在LB/Amp+琼脂板上,因为初始DNA输入量较低。
g. 在32°C下孵育16小时,并在第二天检查菌落。阴性对照板上大量菌落通常表明BbsI消化没有起作用。从加入退火寡核苷酸的平板上挑选4-6个菌落,并分别用每个菌落接种4 ml LB/Amp+培养基。使用一个新的LB/Amp+琼脂板记录所有接种的菌落及其相应的管号。在37°C过夜孵育。
h. 使用如Qiagen QIAprep Spin Miniprep Kit的旋转柱子提取质粒DNA,并使用U6-Forward引物进行测序以鉴定阳性克隆。同时,将步骤A3g中的记录板保持在4°C,直到通过测序确认所有阳性克隆。
i. 从记录板上的阳性克隆菌落接种250 ml LB/Amp+,准备转染级别的质粒。按照制造商的说明培养并提取质粒,使用Qiagen EndoFree Plasmid Maxi Kit确保完全去除内毒素。将纯化的质粒稀释至1 μg/μl或适当浓度,以50-μl分装在无内毒素的TE缓冲液( maxi-prep试剂盒中提供)中,并在使用前储存在-20°C。
C. 人诱导多能干细胞的编辑
在涂有Matrigel的6孔板中培养健康未分化的 人诱导多能干细胞。通常,每个6孔板使用2 ml mTeSR1培养基,每24小时更换一次。每天观察克隆的形态,并注意分化迹象。
当细胞密度达到60-70%(大约4-7天)时,传代培养的iPS细胞。根据技术手册,向每个旧板孔中加入1 ml ReLeSR,并在盖好板盖的情况下孵育3-5分钟。将解离的细胞团稀释,并将1/10至1/20的细胞转移到一个新的预先涂有Matrigel的2 ml mTeSR1的孔中。
详细信息请参考StemCell Technologies提供的技术手册第3部分:在mTeSRTM1中维护人多能干细胞(https://www.stemcell.com/media/files/manual/10000005505-Maintenance_of_Human_Pluripotent_Stem_Cells_mTeSR1.pdf)。
D. 将含有gRNA的Cas9载体和ssODN转染人诱导多能干细胞进行精确基因编辑
在转染前,将诱导多能干细胞播种到60-mm的培养皿中。
a. 作为指导,每个6孔板(60-70%的细胞密度)大约有1 × 106个诱导多能干细胞。另一方面,每个60-mm培养皿需要播种4 × 105-5 × 105个诱导多能干细胞。为了补偿收集和计数过程中的损失,将播种的总诱导多能干细胞孔数乘以1.2。
b. 为每个将要产生的克隆准备好足够数量的60-mm细胞培养级别的培养皿,并预先涂布Matrigel。通常,为每个克隆准备一个培养皿,再加上一个用于评估转染效率的培养皿。
c. 当6孔板中的诱导多能干细胞达到60-70%的细胞密度时,计算转染所需的诱导多能干细胞数量。在消化细胞之前,通过立体显微镜检查细胞形态,并在显微镜下轻轻刮除任何分化的小团。不要使用超过20%的克隆显示出分化迹象的培养物。从培养皿中移除培养基,并添加1 ml Accutase per well。将培养皿放回培养箱中,等待10分钟以消化细胞。
d. 10分钟后,向每个6孔板中的孔加入1 ml室温的mTeSR。轻敲培养板以轻轻地从底部脱落任何剩余的细胞,而不会溢出培养基。将所有孔中的培养基收集到15-ml无菌Falcon管中,使用常规1-ml尖头或玻璃滴管轻轻吹打5-7次,混合均匀并打散团块。
e. 使用血细胞计数器或自动细胞计数器(如ThermoFisher Countess 3)确定细胞密度。此时的细胞密度应为大约3 × 105-6 × 105细胞/ml。同时,将含有细胞的Falcon管保持在室温下;不要将细胞放在冰上。计算收集的细胞总数。
f. 在室温下以200 × g的转速离心5分钟,沉淀细胞。小心移除所有培养基,并加入等体积的新鲜mTeSR1。使用宽口1-ml尖头或玻璃滴管重新悬浮细胞沉淀。再次在室温下以200 × g的转速离心5分钟。
g. 计算用于播种诱导多能干细胞到60-mm培养皿(加一个转染对照培养皿)所需的mTeSR1的最终体积。每个培养皿需要4-5 × 105细胞悬浮在4 ml mTeSR1中。在培养基中加入ROCK抑制剂Y27632,使其最终浓度为5 μM,以提高重新种植后的细胞活力。如果需要进行多个转染(比如,多个培养皿),可能更方便地将部分重新悬浮的细胞转移到一个更大的50-ml Falcon管中,并在转移后添加更多的mTeSR1。从步骤D1f中移除所有培养基,并用含有Y27632的mTeSR1替换。将细胞稀释到所需浓度,并用完全重新悬浮的iPSCs填充每个60-mm培养皿,作为单细胞。在37°C、5% CO2下孵育至少12小时。
转染诱导多能干细胞(iPSCs)与含有gRNA的Cas9载体和单链脱氧核糖核酸(ssODN)。
a. 转染前1小时,检查细胞生长情况,并用4 ml无抗生素的mTeSR1替换培养基。
b. 对于每个60-mm培养皿,在一个1.5-ml管中准备以下混合物。轻轻吹打混合均匀。对于对照培养皿,用pEGFP-N2质粒替换pSpCas9(BB)-2A-Puro构建体。
组件 量(μl)
pSpCas9(BB)-2A-Puro (PX459) V2.0与gRNA插入物,
1 μg/μl 3单链脱氧核糖核酸(ssODN),1 μg/μl 3
Opti-MEM I 减少血清培养基 473
总计 479c. 将21 μl FuGene HD转染试剂(1:3.5 DNA:试剂比例,总共500 μl)加入步骤D2b中的混合物中,并短暂振荡(2-3秒)管子以混合。在室温下孵育最终混合物6分钟。
d. 将DNA:FuGENE:Opti-MEM I混合物加入60-mm培养皿中,并轻轻摇动以混合。不要上下吹打,因为这会干扰转染过程。在37°C、5% CO2下孵育24小时。
E. 选择阳性克隆并用嘌呤霉素(puromycin)评估转染效率
选择阳性克隆
注意:同时阅读步骤D2。
a. 对于所有用pSpCas9(BB)-2A-Puro构建体转染的培养皿,每皿加入4 ml补充有0.5 μg/μl嘌呤霉素的mTeSR1,以选择性地富集转染了带有嘌呤霉素抗性基因的pSpCas9(BB)-2A-Puro构建体的细胞。使用的嘌呤霉素浓度可能需要根据细胞系的特定情况进行优化,因为不同细胞系对嘌呤霉素的耐受水平可能差异很大(范围从0.3到0.7 μg/μl)。在37°C、5% CO2下孵育培养皿24小时。
b. 小心地从培养皿中移除培养基和任何死细胞或细胞碎片。向每皿加入4 ml补充有0.3 μg/μl嘌呤霉素(或根据之前测定确定的浓度)的mTeSR1。在37°C、5% CO2下孵育培养皿另外24小时。
c. 小心地从培养皿中移除培养基和任何死细胞或细胞碎片。从这一时刻起停止嘌呤霉素选择(转染后72小时)。每天向培养皿中加入4 ml mTeSR,并替换培养基。在37°C、5% CO2下孵育培养皿5-10天,直到iPSC克隆达到适合克隆选择的适当大小(直径1.5-2毫米,图3)。

[^]: 图3. 克隆选择的iPSC克隆示例
通过EGFP表达评估转染效率。这一步骤应在步骤D1的同时进行。
a. 对于用pEGFP-N2质粒转染的培养皿,小心地用不含嘌呤霉素的4 ml新鲜mTeSR替换培养基。在37°C、5% CO2下孵育培养皿24小时。
b. 用另外4 ml不含嘌呤霉素的mTeSR替换培养基。在37°C、5% CO2下孵育培养皿另外24小时。
c. 计数几个视野以评估EGFP+细胞的百分比。通过EGFP+细胞的百分比计算的转染效率应至少为40-50%。
分离个体iPSC克隆以鉴定阳性克隆的基因型。
a. 在克隆挑选的那天,准备一个涂有Matrigel的96孔板(称为板A)。在每个孔中加入80 μl含有10 μM ROCK抑制剂Y27632的mTeSR1。准备另一个未涂层的96孔板(称为板B,平面底部孔)。
b. 用新鲜不含ROCK抑制剂的mTeSR1替换60-mm培养皿中的培养基。
c. 将P100或P200移液器设置为50 μl。安装一个P200尖头,并用尖头刮取单个克隆。用移液器吸取刮下的克隆或其碎片(如果在过程中克隆破裂),并将所有内容(50 μl)转移到板A的相应孔中。轻轻吹打3次以打破细胞团。不更换尖头,从板A转移到板B的相应孔中(例如,A1孔到A1孔等)。重复此步骤,直到从60-mm培养皿上挑选出所有感兴趣的克隆。
d. 在室温下,以200 × g的转速离心5分钟,盖好盖子。
e. 将板A保持在37°C,5% CO2下孵育24小时,以使iPSC重新附着到孔中。继续孵育板,并每天更换培养基(每孔100 μl mTeSR1)。板A将在基因型鉴定后用于进一步的克隆扩增。
使用板B进行DNA提取。
a. 小心地从离心机中取出板B,不要摇晃,并将其放在冰上。使用移液器或多通道移液器(设置为200 μl)小心地从孔中移除培养基,不要打扰细胞沉淀。重复此步骤,从板上的所有孔中移除培养基。
b. 在每个含有细胞沉淀的孔中加入16 μl快速DNA提取缓冲液(Lucigen/VWR)。将P10或P50多通道移液器设置为10 μl。小心地吹打5次以混合;尽量减少气泡形成。这一步骤重悬细胞并执行细胞裂解,以便后续使用。
c. 将所有(16 μl)混合的细胞悬液转移到96孔PCR板中,并使用条形盖或薄膜密封。标记板为“提取的DNA”,并注明姓名和日期。如果不在立即使用(1-2周),则应将此步骤的产品储存在-20°C,或者在长期储存的情况下储存在-80°C。设置热循环器如下,并丢弃板B。
Step # Temperature Time Note 1 65°C 15 min 2 68°C 15 min 3 98°C 10 min 4 4°C hold 为Sanger测序准备样本。
a. 使用前一步骤中快速分离的DNA作为模板准备PCR反应混合物。根据以下图表在冰上组装反应,将每个成分乘以样品数量,再乘以1.1以补偿试剂转移过程中的潜在损失。
Component Amount (μl) 10× PCR Buffer 1 10× PCR Enhancer 1 50 mM MgSO4 0.3 10 mM dNTP 0.2 Forward primer (10 μM) 0.2 Reverse primer (10 μM) 0.2 Qiagen HotStarTaq DNA polymerase 0.1 Water, PCR grade 5.5 Total 8.5 b. 在冰上混合所有成分(不包括DNA模板)到一个1.5-ml管中。使用一个新的96孔PCR板,并在每个孔中分配8.5 μl反应混合物,直到所有所需的孔都已填满。随后,在每个孔中加入1.5 μl提取的DNA(总共10 μl)。使用设置为5 μl的P10移液器轻轻吹打,混合均匀,同时避免产生气泡。如果使用多通道移液器,此操作将更为方便。
c. 用条形盖或薄膜密封PCR板的顶部,并标记板为“PCR产物”。设置热循环器如下。退火温度(3)和延伸时间(4)可能需要优化,由操作者根据需要确定。
Step # Temperature Time Note 1 95°C 10 min Taq activation 2 95°C 30 s 3 53°C 90 s Or as appropriate 4 72°C 60 s Or as appropriate 5 / / Go to 2, 40 cycles 6 72°C 10 min Final extension 7 4°C hold d. 在PCR反应运行的同时,设置一个或多个1.25% TAE/TBE琼脂糖凝胶,并准备足够的孔位以容纳至少20个样品的PCR效率检查。
e. 根据以下图表在冰上组装虾碱性磷酸酶(SAP)反应混合物,以从PCR产物中去除单链DNA和dNTPs。将每个成分乘以样品数量,再乘以1.1以补偿试剂转移过程中的潜在损失。
Component Amount (μl) 10× SAP buffer 0.5 Shrimp alkaline phosphatase (SAP) 0.5 Exonuclease I 0.1 Water, PCR grade 3.9 Total 5 f. PCR反应完成后,使用一个新的96孔PCR板,并在每个孔中分配5 μl SAP混合物,直到所有所需的孔都已填满。然后,将标记为“PCR产物”的板上的5 μl(总体积的一半)产品转移到含有SAP混合物的96孔板中的相应孔中(总共10 μl)。轻轻吹打混合均匀,同时避免产生气泡。标记该板为“SAP产物”,并设置热循环器如下。
Step # Temperature Time Note 1 37°C 50 min 2 95°C 15 min 3 4°C hold g. 在SAP反应运行的同时,将1.5 μl 6×上样缓冲液加入“PCR产物”板的选定孔中,并将内容物加载到步骤E4g中准备的TAE/TBE凝胶上。以100 V的电压运行凝胶,至少25分钟,并使用适当的大分子标记物。检查每个通道中的条带的形状、数量和清晰度。拍照并保留记录。
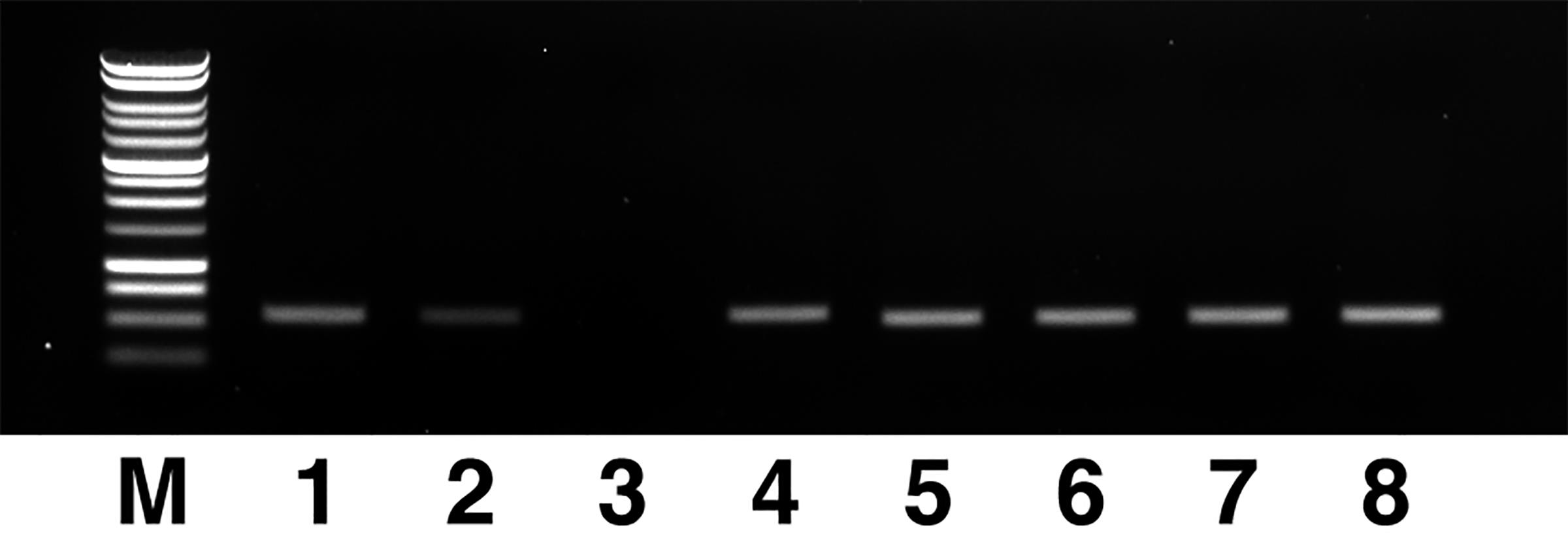
[^图4. PCR产物的琼脂糖凝胶电泳结果示例。]: 第3个条带DNA量太少,无法接受。第2个条带尽管亮度降低,但DNA含量仍然可接受。M:分子标记,1 kb,Promega G5711。
h. 在拍摄PCR反应的凝胶图像后,计算“良好”反应的数量(一个具有正确分子大小和足够DNA量的单条带,根据条带的亮度评估,见图4),并准备Sanger测序的混合物。根据以下表格在冰上组装Sanger反应混合物。仅将“良好”反应的数量乘以每个组分的体积,并将结果再乘以1.1,因为BigDye终止剂试剂很昂贵。
Component Amount (μl) BigDye Terminator V3.1 1 Sequencing primer (10 μM) 1 Water, PCR grade 3 Total 5 i. SAP反应完成后,使用一个新的96孔PCR板,并在每个孔中分配5 μl Sanger测序混合物,直到所有所需的孔都已填满。然后,将标记为“SAP产物”的板上的5 μl(总体积的一半)产品转移到含有Sanger测序混合物的96孔板中的相应孔中(总共10 μl)。轻轻吹打混合均匀,同时避免产生气泡。标记该板为“测序PCR产物”并设置热循环器如下。退火温度(3)可能需要优化,由操作者根据需要确定。
Step # Temperature Time Note 1 96°C 10 s 2 55°C 5 s Or as appropriate 3 60°C 4 min 4 / / Go to 2, 25 cycles 5 4°C hold j. 使用Qiagen DyeEx 2.0 Spin Kit(最适合≤ 16个样品)纯化测序PCR产物。如果DyeEx 2.0 Kit不可用或样品超过16个,跳过步骤E5j,继续执行步骤E5k。
i. 松开顶盖并折断底部的封闭。将离心柱插入收集管中并 centrifuge at 750 × g for 3 min。
ii. 将离心柱转移到一个新的1.5-ml管中,标签上样品名称。从相应的孔中吸取测序PCR产物,并将反应产物缓慢地直接倒在倾斜的凝胶床表面中心。再次 centrifuge at 750 × g for 3 min。
iii. 丢弃离心柱,并将1.5-ml管放入离心真空设备(如SpeedVac)中,30-60分钟,直到所有液体蒸发。在真空过程中不要打开加热。干燥后,向管中加入15 μl测序级甲酰胺,并轻轻吹打10次以溶解DNA。继续执行步骤E5kvii。k. 通过异丙醇沉淀纯化测序PCR产物。
注意:此步骤至关重要,操作时应小心谨慎。 i. 向每个孔中添加40 μl 75%的异丙醇,并轻轻吹打5次以混合。如果使用多通道移液器,此操作将更为方便。
ii. 在室温下孵育20分钟,然后在室温(20°C)下的摇摆式离心机中以3,000 × g的转速离心30分钟。如果可能,开启离心机的冷却功能,因为离心机内的空气可能会迅速变热。
iii. 小心地从离心机中取出板,不要摇晃。将板轻轻倒置在3-4层纸巾上,以倒出异丙醇。重复此过程一次,使用另一组新的3-4层纸巾,并保持板倒置10秒,以尽可能去除尽可能多的异丙醇。不要使用移液器或真空吸管从孔中移除任何残留的异丙醇。
iv. 慢慢地将96孔板翻转回来,并向每个孔中加入150 μl 75%的异丙醇以洗涤沉淀。
重要:不要混合!如果使用多通道移液器,此操作将更为方便。再次以3,000 × g的转速离心10分钟。 v. 小心地将板倒置在纸巾堆上,如步骤E5kiii中所述,以倒出异丙醇。保持板倒置(!),小心地将它转移到新的纸巾堆上,并再次以300 × g(!)的转速离心1分钟,以消除残留的异丙醇。
vi. 小心地从离心机中取出96孔板,并将其翻转回来。向每个孔中加入15 μl测序级甲酰胺,并轻轻吹打5次以混合。避免在井内形成气泡。
vii.用粘合膜密封板,并在热循环器上于95°C变性2分钟。立即将板放在冰上,并孵育2分钟。
viii. 以2,000 × g的转速离心1分钟。现在该板已准备好在 ABI 310/3730/3730xl/3500测序仪上进行Sanger测序注射。
F. 扩增具有所需基因型的阳性克隆。
使用ApE或ABI GeneMapper检查测序光谱文件,以识别需要扩增的阳性候选克隆(见第A节,图1和图5)。扩增的iPSCs将用于进一步的单细胞亚克隆,以获得纯合子克隆。
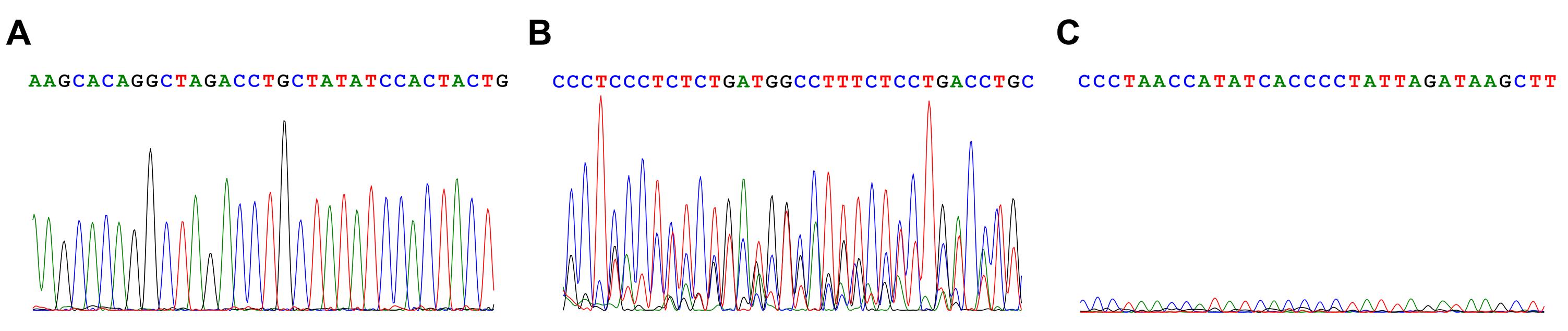
[^]: 图5. 良好和质量差的Sanger测序光谱示例。
a. 当 colonies 在 Plate A 中达到适当的大小(1.5-2毫米直径或60-70%的孔内覆盖率)(从步骤E3a),通过首先从每个孔中移除所有培养基来消化iPSC克隆。轻轻添加50 μl ReLeSR到每个孔中,快速抽吸,并在室温下站立5分钟。
b. 向每个孔中添加100 μl mTeSR1,并小心地用常规P200移液器尖头从井底松动细胞团。将每个井的内容转移到一个涂有Matrigel的4孔或24孔板中的一个井中,并添加含5 μM ROCK抑制剂的mTeSR1,最终体积为500 μl。在培养箱中继续培养,37°C,5% CO2,每天更换mTeSR1,直到细胞在井内达到60-70%的覆盖率。
单细胞亚克隆以获得纯合子细胞系。
a. 小心地从培养皿中移除培养基,然后向每个孔中加入400 μl Accutase,孵育10分钟以从底部分离细胞。使用P1000尖头轻轻吹打5次,制成单细胞悬液,然后加入另外400 μl mTeSR1以中和Accutase。取10 μl悬液使用台盼蓝法计数细胞密度,并将剩余的细胞以250 × g的转速离心5分钟。
b. 移除上清液,并在1 ml含有5 μM ROCK抑制剂Y-27632的mTeSR1中重悬细胞。分离出相当于2000个活细胞的体积(根据前一步骤确定),并在含有5 μM ROCK抑制剂Y27632的4 ml mTeSR1中重新种植到60-mm Matrigel涂层的培养皿中。继续培养任何剩余的细胞作为备份,培养条件为37°C,5% CO2。
c. 在重新种植后48小时,用ROCK抑制剂-free mTeSR1替换60-mm培养皿中一半的培养基。在培养箱中继续培养,37°C,5% CO2,每天更换mTeSR1,直到克隆大小达到1.5-2毫米。重复步骤E3中的克隆挑选程序。从每个60-mm培养皿中挑选约30个克隆。对选定的克隆重复步骤E4-E5中的基因型鉴定程序。
d. 一旦通过Sanger测序识别出纯合子克隆,重复步骤F1,将每个克隆从96孔板转移到Matrigel涂层的4/24孔板的2个(!)孔中,直到细胞密度达到60-70%。继续监测细胞生长,因为不同克隆在这个阶段的生长速度可能不同。
e. 从培养皿中移除培养基,并向每个孔中加入400 μl ReLeSR。从孔中移除ReLeSR,并在室温下孵育5分钟。
f. 预先涂布足够数量的6孔板Matrigel,并向每个孔中加入2 ml mTeSR1。向每个用ReLeSR处理的4/24孔板的孔中加入300 μl mTeSR1,并使用宽口移液器尖头轻轻吹打5次以分离细胞团块。将100 μl细胞团块悬液分到6孔板(总共3个孔)的每个孔中,并轻轻摇动混合。为每个克隆准备4-6个孔。继续培养,直到细胞密度达到60-70%。
冻存细胞系以进行冷冻保存。
a. 密切观察6孔板中的细胞生长,直到细胞密度达到60-70%。建议同一板上的所有孔在一次处理中完成。
b. 向每个6孔板孔中加入1 ml ReLeSR。从孔中移除ReLeSR,并在37°C、5% CO2的培养箱中孵育3-5分钟。
c. 向每个用ReLeSR处理的6孔板孔中加入1 ml室温mTeSR1,并通过坚定地轻敲培养板来松动细胞。将松动的细胞悬液转移到一个无菌的1.5-ml Eppendorf管中,并在室温下以200 × g的转速离心5分钟。
d. 小心地尽可能多地移除上清液,同时不要触及底部的细胞沉淀。向Eppendorf管中加入1 ml室温mFreSR,并使用宽口移液器尖头小心地重新悬浮细胞沉淀,通过吹打5次来混合。将管中的内容物转移到一个预先标记的冷冻管中。确认管盖已牢固密封,并将其放入Mr Frosty冷冻容器中。通常,为每个克隆准备尽可能多的管(通常为4-6管,如步骤F2d-F2f中所确定)。重复此步骤,直到所有细胞都转移到冷冻管中。
e. 将装有冷冻管的Mr Frosty冷冻容器放入-80°C的冷冻器中至少24小时(但不超过72小时)以冷冻内容物。将所有试管转移到液氮中进行长期保存。保持详细记录。
数据分析
Sanger测序结果的光谱分析
- 成功识别和分离精确编辑的纯合子克隆,在很大程度上依赖于对Sanger测序结果的适当分析。作为指导,首先去除信号强度低、异常模式(例如,当连续序列中出现相同类型的核苷酸碱基时,合并成一个大峰而不是个体峰,见图5)。随后,通过检查所需SNP位点的基因型来识别阳性克隆。
- 评估gRNA和ssODN的效率
由于ssODN的整合和随后的精确SNP编辑需要CRISPR/Cas9介导的DNA切割优先发生,因此需要独立评估CRISPR/Cas9介导的DNA切割和ssODN整合的效率。通常,由于大多数CRISPR/Cas9介导的DNA断裂没有适当修复并产生插入缺失(indels),因此生成的包含indel的克隆百分比(见图1)可以作为实验中使用的gRNA序列效率的指标。包含indel的克隆百分比对于成功的gRNA序列设计约为60-80%。ssODN的整合相对稳定,大约占CRISPR/Cas9介导的DNA切割事件的5%。如果阳性克隆的数量低,可以从Sanger测序结果中分别计算两个事件的可能性,以确定哪个寡核苷酸序列可能是原因并需要重新设计。
References
- Forrest, M. P., Zhang, H., Moy, W., McGowan, H., Leites, C., Dionisio, L. E., Xu, Z., Shi, J., Sanders, A. R., Greenleaf, W. J., Cowan, C. A., Pang, Z. P., Gejman, P. V., Penzes, P. and Duan, J. (2017). Open Chromatin Profiling in hiPSC-Derived Neurons Prioritizes Functional Noncoding Psychiatric Risk Variants and Highlights Neurodevelopmental Loci. Cell Stem Cell 21(3): 305-318 e308.
- Forsyth, N. R., Musio, A., Vezzoni, P., Simpson, A. H., Noble, B. S. and McWhir, J. (2006). Physiologic oxygen enhances human embryonic stem cell clonal recovery and reduces chromosomal abnormalities. Cloning Stem Cells 8(1): 16-23.
- Hendel, A., Kildebeck, E. J., Fine, E. J., Clark, J., Punjya, N., Sebastiano, V., Bao, G. and Porteus, M. H. (2014). Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep 7(1): 293-305.
- Howden, S. E., Maufort, J. P., Duffin, B. M., Elefanty, A. G., Stanley, E. G. and Thomson, J. A. (2015). Simultaneous Reprogramming and Gene Correction of Patient Fibroblasts. Stem Cell Reports 5(6): 1109-1118.
- Miyaoka, Y., Chan, A. H., Judge, L. M., Yoo, J., Huang, M., Nguyen, T. D., Lizarraga, P. P., So, P. L. and Conklin, B. R. (2014). Isolation of single-base genome-edited human iPS cells without antibiotic selection.Nat Methods 11(3): 291-293.
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A. and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11): 2281-2308.
- Tidball, A. M., Swaminathan, P., Dang, L. T. and Parent, J. M. (2018). Generating Loss-of-function iPSC Lines with Combined CRISPR Indel Formation and Reprogramming from Human Fibroblasts. Bio-protoc**ol 8(7): 2794.
- Zhang, S., Moy, W., Zhang, H., Leites, C., McGowan, H., Shi, J., Sanders, A. R., Pang, Z. P., Gejman, P. V. and Duan, J. (2018). Open chromatin dynamics reveals stage-specific transcriptional networks in hiPSC-based neurodevelopmental model. Stem Cell Res 29: 88-98.
- Zhang, S., Zhang, H., Zhou, Y., Qiao, M., Zhao, S., Kozlova, A., Shi, J., Sanders, A. R., Wang, G., Luo, K., Sengupta, S., West, S., Qian, S., Streit, M., Avramopoulos, D., Cowan, C. A., Chen, M., Pang, Z. P., Gejman, P. V., He, X. and Duan, J. (2020). Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants.Science 369(6503): 561-565.
- 本文作者: Anderson
- 本文链接: http://nikolahuang.github.io/2024/03/14/CRISPR-Cas9介导的人iPSC细胞系SNP精确编辑/
- 版权声明: 转载请注明出处,谢谢。