

WH Wenjie Han,HL Haojun Liang ![]() ,JB Jianqiang Bao
,JB Jianqiang Bao ![]()
第一次发布: 2023年10月20日第13卷第20期 DOI: 10.21769/BioProtoc.4853 评审: Xin Xu 匿名评审
My notes:
这项研究主要介绍了一种名为“LOCK”(Long dsDNA with 3′-Overhangs mediated CRISPR Knock-in)的基因组编辑技术,旨在实现高效的大DNA片段敲入。其关键创新点包括:
3’悬垂的双链DNA(odsDNA)供体:在双链DNA供体的3’端设计一段单链DNA悬垂序列,可显著提高基因组敲入的效率。
PCV2连接:通过设计一个含有PCV2蛋白识别序列的连接物,将odsDNA连接到Cas9-PCV2融合蛋白上,进一步提高了敲入效率。
长DNA片段敲入:通过使用odsDNA/Cas9-PCV2/sgRNA复合物,研究实现了1-3kb长DNA片段的高效敲入,敲入效率显著高于传统dsDNA供体。
多种基因位点的敲入验证:研究在HEK293T细胞的3个不同基因位点进行了敲入验证,证明了该方法的有效性和通用性。
优化PCR扩增条件:研究优化了PCR扩增条件,包括使用高保真酶、调整循环参数等,以确保获取高质量的双链DNA供体。
简要的实验步骤概括为:
目标序列选择和引物设计:使用在线工具CHOPCHOP选择目标序列,并在PCR引物中设计同源臂序列。
双链DNA供体和odsDNA供体制备:通过PCR扩增双链DNA供体,并通过引物中的硫代磷酸修饰制备odsDNA供体。
Cas9和Cas9-PCV2蛋白表达纯化:在大肠杆菌中表达Cas9和Cas9-PCV2融合蛋白,并进行纯化。
sgRNA合成和纯化:合成针对目标基因的sgRNA,并使用试剂盒进行纯化。
RNP复合物制备:将odsDNA连接到Cas9-PCV2上,制备RNP复合物。
核转染RNP复合物:将RNP复合物核转染到HEK293T细胞中,实现基因组敲入。
敲入效率检测:通过流式细胞仪和PCR检测敲入效率。
验证不同长度的DNA片段敲入:比较不同长度DNA片段的敲入效率。
Abstract
在全球分子生物学和细胞生物学实验室中,高效且精准的基因组编辑技术极为重要。尽管目前碱基编辑器(base editors)和主编辑器(prime editors)等精确基因组编辑工具已取得快速发展,但成本效益高、适用于长DNA片段的高效敲入(KI)技术仍不可或缺。我们之前的研究利用微同源性介导的末端连接高效修复双链断裂(DSB),展示了结合CRISPR-Cas9技术的一种特殊设计的带有50个核苷酸同源性臂(HA)的3′-悬垂双链DNA(odsDNA)供体,可实现高效的外源DNA敲入。odsDNA供体的3′-悬垂长度可通过五个连续的硫代磷酸(PT)修饰来调控。本指南详细介绍了执行LOCK(利用3′-悬垂的长双链DNA进行CRISPR敲入)方法的步骤,该方法适用于在哺乳动物细胞中进行基因大小(约1-3 kb)的敲入。
Graphical overview
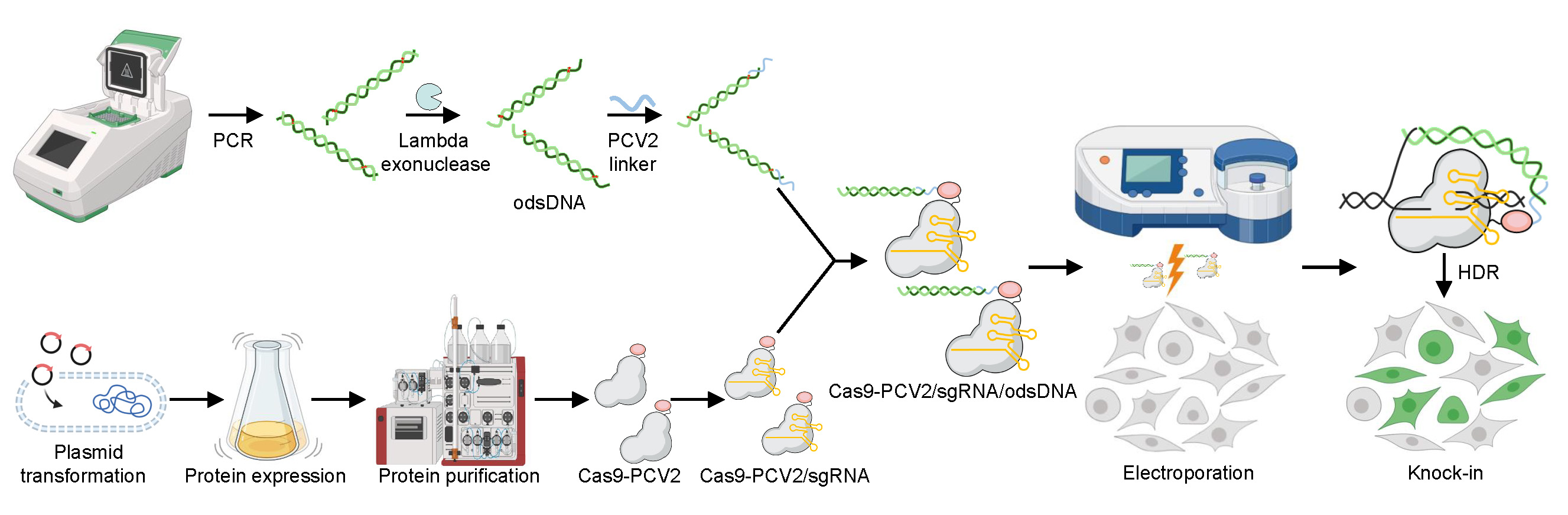
CRISPR及其衍生技术的出现极大地扩展了生物医学研究和创新生物技术开发领域中科学家进行基因组操作的“工具箱”。与高度依赖专门工程化的蛋白阅读器来特异性识别目标基因组DNA(gDNA)序列的ZFN或TALEN方法相比,CRISPR-Cas9方法仅需要一个单一的crRNA(约20个核苷酸),它能独特地识别并与特定的gDNA位点配对(Hsu等人,2014年)。这一改进使得基因组编辑技术因其低成本和简化的程序而在任何个人实验室都可采用。
然而,众所周知,大型供体DNA序列精确整合到宿主基因组依赖于同源定向修复(HDR),这在哺乳动物细胞中发生的频率较低。此外,Cas9切割引起的双链断裂(DSB)位点可能发生在不期望的基因组位点上(脱靶),这可能导致意外的DSB修复结果,例如插入/缺失(indels)和易位,这对细胞是有害的。因此,在最小化脱靶效应的同时提高精确靶向效率至关重要(Doudna,2020年)。
在过去的十年里,科学家们在提高精确CRISPR-Cas9敲入(KI)效率方面取得了巨大成就。供体DNA序列的磷酸骨架具有高度负电荷,这自然阻碍了它们穿过细胞膜,导致外源DNA供体在局部DSB位点无法获得。这被认为是导致HDR介导KI效率低下的原因之一。因此,迄今为止,已经设计出了多种方法来促进外源修复DNA供体的核内递送或结构稳定性(Yu等人,2020年)。例如,通过腺相关病毒(AAV)载体、Strep-biotin标记的连接(Gu等人,2018年)或染色质包装(Cruz-Becerra和Kadonaga,2020年)可以促进DNA供体的核内进入。
另一方面,众所周知,虽然经典的非同源末端连接(c-NHEJ)修复在CRISPR-Cas9诱导的DSB位点更为频繁,但通过人为操纵细胞环境,修复途径可以偏向精确的HDR修复结果(Maruyama等人,2015年)。事实上,越来越多的例子表明,长单链DNA(lssDNA,oligo)作为外源HDR供体,在HDR效率和脱靶效应方面比传统的双链DNA(dsDNA)供体具有更大的优势(Quadros等人,2017年;Shy等人,2023年)。利用dsDNA和ssDNA供体的独特优势,我们之前的研究表明,单一的杂交“3′-悬垂dsDNA”供体,称为odsDNA,已被证明可以显著提高HDR效率,最多提高5倍,而脱靶效应较低(Han等人,2023年)。我们将这种方法简称为“LOCK”(Long dsDNA with 3′-Overhangs mediated CRISPR Knock-in)。在本协议中,我们描述了在任何个人实验室操作的这一简单技术的步骤。
实验操作细则:
A. 为了在HEK293T细胞的不同基因组位点上实现绿色荧光蛋白的敲入(KI),需要准备dsDNA和带有3′-悬垂的dsDNA(odsDNA)作为DNA供体
1.选取了Lamin A/C、GAPDH和AAVS1位点的目标序列作为示例。
a. 在这些供体DNA序列中,被插入的序列两侧各有50个核苷酸的同源性臂。
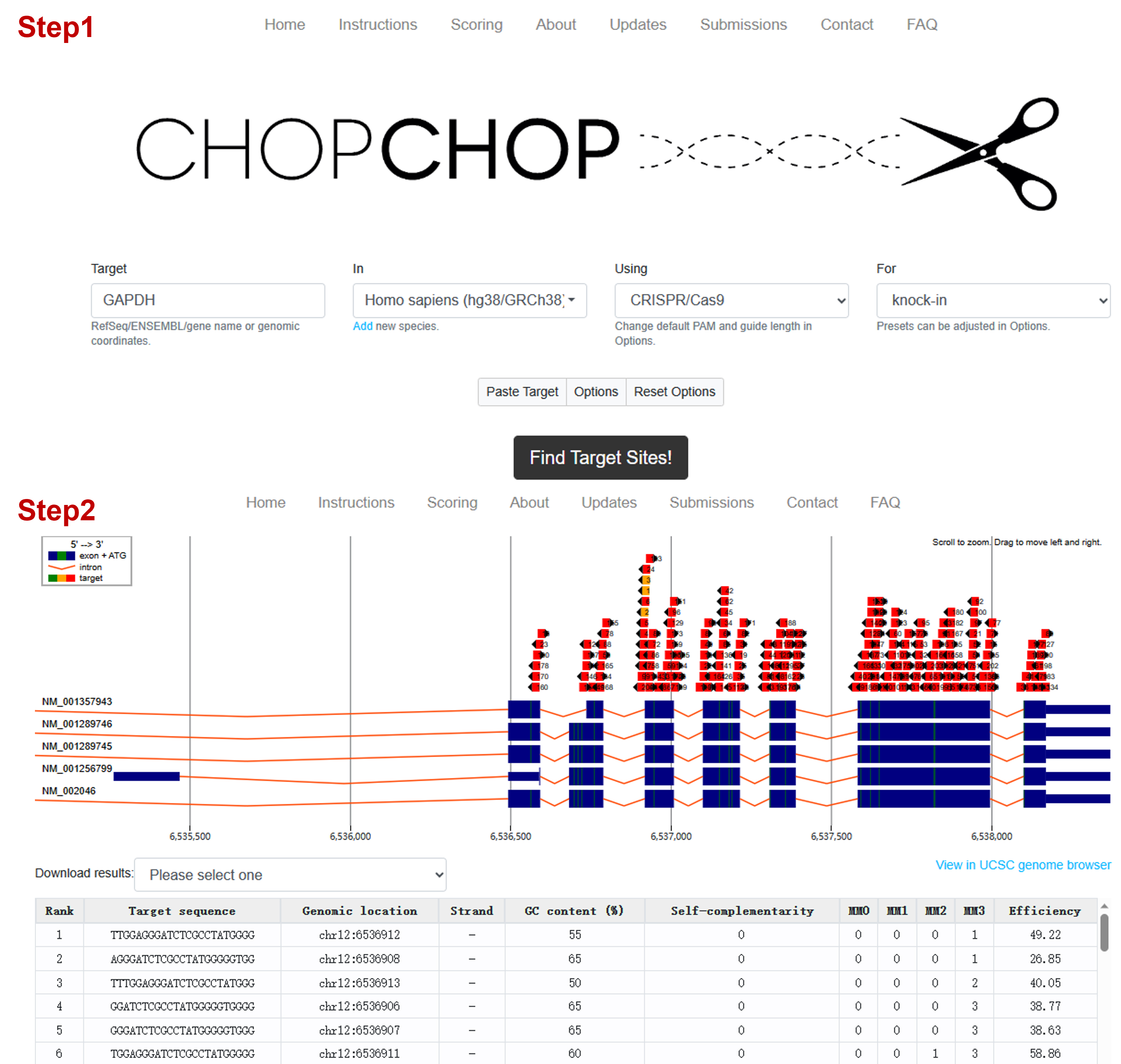
Note: 目标序列是使用在线工具CHOPCHOP选择的,选择的标准是序列中错配较少且得分较高。一个具体的示例已在图1中展示。
[^Fig1]: Screenshot example of the CHOPCHOP website when selecting the target sequence
b. 在本研究中,我们采用了长度为1,010和2,500个碱基对的基因片段作为DNA供体。这些片段按照标准的克隆程序被插入到质粒中,这些质粒包括了必要的启动子区域和EGFP(增强型绿色荧光蛋白)序列。在这些供体DNA序列中,用于同源定向修复(HDR)的50个核苷酸同源性臂(HA)序列已被下划线标出。具体的模板序列详见表1。
| Lamin A/C locus donor sequence (EF-1α core promoter-EGFP 1110bp) |
|---|
| ctttggtttttttcttctgtatttgtttttctaagagaagttattttctataggtcttgaaaggagtgggtcaattggctccggtgcccgtcagtgggcagagcgcacatcgcccacagtccccgagaagttggggggaggggtcggcaattgatccggtgcctagagaaggtggcgcggggtaaactgggaaagtgatgtcgtgtactggctccgcctttttcccgagggtgggggagaaccgtatataagtgcagtagtcgccgtgaacgttctttttcgcaacgggtttgccgccagaacacaggaagcttgccaccatggtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaagtaagactctggtcagagatacctcagtggttttatactgaaggaaaaacacaagcaaaaaaaaaaaaaaagca |
| *GAPDH* locus donor sequence (EF-1α core promoter-EGFP 1110bp) |
| atggcctccaaggagtaagacccctggaccaccagccccagcaagagcactaggtcttgaaaggagtgggtcaattggctccggtgcccgtcagtgggcagagcgcacatcgcccacagtccccgagaagttggggggaggggtcggcaattgatccggtgcctagagaaggtggcgcggggtaaactgggaaagtgatgtcgtgtactggctccgcctttttcccgagggtgggggagaaccgtatataagtgcagtagtcgccgtgaacgttctttttcgcaacgggtttgccgccagaacacaggaagcttgccaccatggtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaagtaagactctggtcagagatacctaagaggaagagagagaccctcactgctggggagtccctgccacactcagt |
| *AAVS1* locus donor sequence (EF-1α core promoter-EGFP 1110bp) |
| gttctgggtacttttatctgtcccctccaccccacagtggggccactaggtaggtcttgaaaggagtgggtcaattggctccggtgcccgtcagtgggcagagcgcacatcgcccacagtccccgagaagttggggggaggggtcggcaattgatccggtgcctagagaaggtggcgcggggtaaactgggaaagtgatgtcgtgtactggctccgcctttttcccgagggtgggggagaaccgtatataagtgcagtagtcgccgtgaacgttctttttcgcaacgggtttgccgccagaacacaggaagcttgccaccatggtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaagtaagactctggtcagagatacctgacaggattggtgacagaaaagcccccatccttaggcctcctccttccta |
| *Lamin A/C* locus donor sequence (EF-1α promoter-EGFP 2600bp) |
| ctttggtttttttcttctgtatttgtttttctaagagaagttattttctatgcccggcgagagatcacgtggggcgcggaggcggtgctgctggggcacggccgtccagcctcggcggccatatttttgaggggctgttcatctcgttcacacgctctgtccgccatgtttgtgagtggaagcgccattaccccttcaagcgactgaaggctgcagggcctctggtggcccgcatggggagaccagacccgccaggcccgcctttccgcactcagtccgggcttactttattttgtgagacagggtctcgcctagaggctccggtgcccgtcagtgggcagagcgcacatcgcccacagtccccgagaagttggggggaggggtcggcaattgaaccggtgcctagagaaggtggcgcggggtaaactgggaaagtgatgtcgtgtactggctccgcctttttcccgagggtgggggagaaccgtatataagtgcagtagtcgccgtgaacgttctttttcgcaacgggtttgccgccagaacacaggtaagtgccgtgtgtggttcccgcgggcctggcctctttacgggttatggcccttgcgtgccttgaattacttccacgcccctggctgcagtacgtgattcttgatcccgagcttcgggttggaagtgggtgggagagttcgaggccttgcgcttaaggagccccttcgcctcgtgcttgagttgaggcttggcctgggcgctggggccgccgcgtgcgaatctggtggcaccttcgcgcctgtctcgctgctttcgataagtctctagccatttaaaatttttgatgacctgctgcgacgctttttttctggcaagatagtcttgtaaatgcgggccaagatctgcacactggtatttcggtttttggggccgcgggcggcgacggggcccgtgcgtcccagcgcacatgttcggcgaggcggggcctgcgagcgcggccaccgagaatcggacgggggtagtctcaagctggccggcctgctctggtgcctggcctcgcgccgccgtgtatcgccccgccctgggcggcaaggctggcccggtcggcaccagttgcgtgagcggaaagatggccgcttcccggccctgctgcagggagctcaaaatggaggacgcggcgctcgggagagcgggcgggtgagtcacccacacaaaggaaaagggcctttccgtcctcagccgtcgcttcatgtgactccacggagtaccgggcgccgtccaggcacctcgattagttctcgagcttttggagtacgtcgtctttaggttggggggaggggttttatgcgatggagtttccccatactgagtgggtggagactgaagttaggccagcttggcacttgatgtaattctccttggaatttgccctttttgagtttggatcttggttcattctcaagcctcagacagtggttcaaagtttttttcttccatttaaggtgtcgtgaaaactaccccaagctggcctctgaggccaccatggctgtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaagtaaaagcttggggatcaattctctagagctcgctgatcagcctcgactgtgccttctagttgccagccatctgttgtttgcccctcccccgtgccttccttgaccctggaaggtgccactcccactgtcctttcctaataaaatgaggaaattgcatcgcattgtctgagtaggtgtcattctattctggggggtggggtggggcaggacagcaagggggaggattgggaagacaatagcaggcatgctggggatgcggtgggctctatggcttctgaggcggaaagaaccagctgggcccagtggttttatactgaaggaaaaacacaagcaaaaaaaaaaaaaaagca |
[^table1.Sequences of the target insert in donor plasmids used as PCR templates]:
在进行DNA供体PCR引物设计时,我们遵循以下原则:
对于dsDNA供体引物,我们在合成的前向和反向引物的5′端仅设计两个50个核苷酸的同源性臂(HAs)。这样的设计有助于提高同源定向修复(HDR)的效率,但同时较长的HA序列也可能增加PCR的难度。
2.DNA供体(donor) PCR引物设计和合成
对于odsDNA供体引物的设计,我们采取以下步骤:
a. 与dsDNA类似,在合成的前向和反向引物的5′端分别设计两条50个核苷酸的同源臂序列(HAs)。
注意:较长的HA序列通常导致较高的HDR效率,但也可能增加PCR难度。
b. 设计odsDNA供体引物的原则:
i. 同样,在合成的前向和反向引物的5′端分别设计两个50个核苷酸的HA序列。
ii. 根据需要,在合成的50 nt引物的指定位置指定5个连续的硫代磷酸酯修饰。“*”表示硫代磷酸酯(PT)修饰。小写字母是HA序列,大写字母描绘外源性KI序列。为了节省引物合成的成本,我们使用第一个PCR产物作为模板; 不需要长引物。引物序列如表2所示。
Table 2. Synthesis of PCR primer sequences for dsDNA and odsDNA donors
| Lamin A/C locus | EF-1α core promoter-EGFP (1,110 bp) primers or****EF-1α promoter-EGFP-ploy A signal (2,600 bp) primers (5′→3′) |
|---|---|
| L-50-F | ctttggtttttttcttctgtatttgtttttctaagagaagttattttctaTAGGTCTTGAAAGGAGTGGG |
| L-50-R | tgctttttttttttttttgcttgtgtttttccttcagtataaaaccactgAGGTATCTCTGACCAGAGTC |
| L-30S-F | ctttggtttttttcttctgtatttgtttttctaag*agaagttattttcta |
| L-30S-R | tgctttttttttttttttgcttgtgtttttccttc*agtataaaaccactg |
| L-20S-F | ctttggtttttttcttctgtatttg*tttttctaagagaag |
| L-20S-R | tgctttttttttttttttgcttgtg*tttttccttcagtat |
| L-15S-F | ctttggtttttttcttctgt*atttgtttttctaag |
| L-15S-R | tgctttttttttttttttgc*ttgtgtttttccttc |
| L-10S-F | ctttggtttttttct*tctgtatttgttttt |
| L-10S-R | tgctttttttttttt*tttgcttgtgttttt |
| L-5S-F | ctttggtttt*tttcttctgtatttg |
| L-5S-R | tgcttttttt*ttttttttgcttgtg |
| GAPDH locus | EF-1α core promoter-EGFP primers |
| G-50-F | atggcctccaaggagtaagacccctggaccaccagccccagcaagagcacTAGGTCTTGAAAGGAGTGGG |
| G-50-R | actgagtgtggcagggactccccagcagtgagggtctctctcttcctcttAGGTATCTCTGACCAGAGTC |
| G-30S-F | atggcctccaaggagtaagacccctggaccaccag*ccccagcaagagcac |
| G-30S-R | actgagtgtggcagggactccccagcagtgagggt*ctctctcttcctctt |
| G-20S-F | atggcctccaaggagtaagacccct*ggaccaccagcccca |
| G-20S-R | actgagtgtggcagggactccccag*cagtgagggtctctc |
| G-15S-F | atggcctccaaggagtaaga*cccctggaccaccag |
| G-15S-R | actgagtgtggcagggactc*cccagcagtgagggt |
| G-10S-F | atggcctccaaggag*taagacccctggacc |
| G-10S-R | actgagtgtggcagg*gactccccagcagtg |
| G-5S-F | atggcctcca*aggagtaagacccct |
| G-5S-R | actgagtgtg*gcagggactccccag |
| AAVS1 locus | EF-1α core promoter-EGFP primers |
| A-50-F | gttctgggtacttttatctgtcccctccaccccacagtggggccactaggTAGGTCTTGAAAGGAGTGGG |
| A-50-R | taggaaggaggaggcctaaggatgggggcttttctgtcaccaatcctgtcAGGTATCTCTGACCAGAGTC |
| A-30S-F | gttctgggtacttttatctgtcccctccaccccac*a |
| A-30S-R | taggaaggaggaggcctaaggatgggggcttttct*g |
| A-20S-F | gttctgggtacttttatctgtcccc*t |
| A-20S-R | taggaaggaggaggcctaaggatgg*g |
| A-15S-F | gttctgggtacttttatctg*t |
| A-15S-R | taggaaggaggaggcctaag*g |
| A-10S-F | gttctgggtactttt*atctg |
| A-10S-R | taggaaggaggaggc*ctaag |
| A-5S-F | gttctgggta*cttttatctg |
| A-5S-R | taggaaggag*gaggcctaag |
| Lamin A/C loucs | EF-1α promoter-EGFP-ploy A signal (2,600 bp) primers for esgRNA |
| L-12S-F | ctttggtttttttcttc*tgtatttgtttttctaagag |
| L-12S-R | attgagatagatgagatagatgctttttttttttttt*t |
c. 引物由上海桑戈生物科技有限公司合成。每份引物的干粉首先使用低EDTA TE缓冲液(10 mM Tris和0.2 mM EDTA,pH 8.0)稀释至100 μM(储存液),然后使用无核酸酶的ddH2O稀释至10 μM(工作液)。
d. 供体DNA的PCR扩增:
i. 在400 μL主混合液体积中设置以下PCR组装反应。试剂和体积显示在表3中。涡旋5秒,快速离心,并使用0.2 mL的八管条带制备50 μL分装。
Table 3. Components of PCR reaction for DNA donor
| Reagent | Volume |
|---|---|
| Q5 High-Fidelity 2× Master Mix | 200 μL |
| 10 μM forward primer | 20 μL |
| 10 μM reverse primer | 20 μL |
| Template DNA (plasmids or PCR product) | 80 ng |
| Nuclease-free water | to 400 μL |
ii. 根据表3中列出的顺序,将所有反应成分加入到主混合液中。之后,将400 μL的反应溶液分装成50 μL的小份,使用0.2 mL的八连管条带进行分装。
iii. 根据表4中提供的循环参数,进行组装PCR反应。这些参数包括变性、退火和延伸的温度以及在每个阶段所需的时间,这些参数对于确保PCR反应的准确性和效率至关重要。
Table 4. PCR reaction for DNA donor preparation
| Cycle step | Temperature | Time | Cycles |
|---|---|---|---|
| Initial denaturation | 98 °C | 30 s | 1× |
| Denaturation | 98 °C | 5 s | 35× |
| Annealing | 65 °C | 15 s | |
| Final extension | 72 °C | 1 min/2 min 20 s | 1× |
| Hold | 4 °C | Hold | 1× |
3.使用GeneJET PCR纯化试剂盒对PCR DNA供体进行纯化
a. 将等体积的结合缓冲液加入已完成的PCR混合物中(对于每400 μL的反应混合物,加入400 μL的结合缓冲液)。轻轻上下吹打混合均匀。
b. 将步骤A3a中的不超过800 μL的溶液转移到GeneJET纯化柱上。在12,000× g的条件下离心60秒,并丢弃流穿液。
c. 向GeneJET纯化柱中加入700 μL的洗涤缓冲液。再次在12,000× g的条件下离心60秒,并丢弃流穿液。然后将纯化柱放回收集管中。
d. 将空的GeneJET纯化柱再次离心1分钟,以确保完全去除残留的洗涤缓冲液。
e. 将GeneJET纯化柱转移到一个干净的1.5 mL微量离心管中。在GeneJET纯化柱膜的中央加入25 μL的无核酸酶水,并在12,000× g的条件下离心1分钟。为了提高产量,可以将废液加入GeneJET纯化柱膜上,并重复操作。
f. 使用NanoDrop 2000测量DNA浓度。在每次使用前后,使用Kimwipes一次性擦拭纸擦拭NanoDrop 2000。将纯化的双链DNA供体储存在-20℃的冰箱中。
注意:经过硫代磷酸(PT)修饰的DNA产物(odsDNA前体)需要经过Lambda外切酶消化处理,以制备用于基因敲入的odsDNA供体模板。
4.准备odsDNA 供体
a. 在一个干净的1.5 mL微量离心管中,准备一个300 μL体积的消化反应。表5中列出了所需的试剂和各自的体积。
Table 5. Components to odsDNA donor digestion reaction
| Reagent | Volume |
|---|---|
| Lambda exonuclease reaction buffer (10×) | 30 μL |
| Lambda exonuclease | 6 μL |
| Purified odsDNA precursor (3.f) | 18 μg |
| Nuclease-free H2O | to 300 μL |
b. 在干浴孵化器中,将温度设置为37°C,并让消化反应持续进行60分钟。
5.为了纯化经过Lambda外切酶消化处理后的带有3′-悬垂的双链DNA(odsDNA)供体,我们按照以下步骤使用GeneJET PCR纯化试剂盒进行操作:
a. 将等体积的结合缓冲液加入PCR混合物中(每300 μL反应混合物加入300 μL结合缓冲液),并通过轻轻上下吹打混合均匀。
b. 将步骤A5a中的不超过600 μL的溶液转移到GeneJET纯化柱上。在离心机中以12,000× g的速率离心60秒,并丢弃流穿液。
c. 向GeneJET纯化柱中加入700 μL的洗涤缓冲液。再次以12,000× g的速率离心60秒,并丢弃流穿液。然后将纯化柱放回收集管中。
d. 将空的GeneJET纯化柱再次离心1分钟,以确保完全去除任何残留的洗涤缓冲液。
e. 将GeneJET纯化柱转移到一个干净的1.5 mL微量离心管中。在GeneJET纯化柱膜的中央加入25 μL的无核酸酶水,并在离心机中离心1分钟。为了提高产量,可以将洗脱液加入GeneJET纯化柱膜上,并再次离心。
f. 使用NanoDrop 2000测量DNA浓度。将纯化的odsDNA供体储存在-20℃的冰箱中。
B. 在大肠杆菌细胞中表达和纯化Cas9和Cas9-PCV2融合蛋白
将能容纳的E. coli(Rosetta)细胞与表达质粒pET28b-3NLS-Cas9-3NLS-His进行转化,并在含有卡那霉素的LB平板上培养过夜。
第二天,从含有卡那霉素的LB平板上挑选一个菌落,转移到25 mL的新鲜LB培养基(含卡那霉素)中,然后在37℃下以200 rpm的速度在摇床上培养过夜。
将10 mL过夜培养物接种到含有1L新鲜TB培养基(含卡那霉素)的烧瓶中,并在37℃下以200 rpm摇动。培养细菌,直至OD600达到约0.6–0.8。
注意:TB培养基的最佳生长时间较长,需要在IPTG诱导前OD600超过1.0–1.5。在冰上或冷室中冷却20分钟后,加入IPTG至终浓度为0.2 mM。然后在18℃下继续摇动培养过夜(18–20小时)。
第二天,离心培养液并称量沉淀。用缓冲液A以约6 mL/g的比例重新悬浮细胞。
向每50 mL细菌悬浮液中加入50 μL β-巯基乙醇。进行超声处理:设置35%振幅,1.5秒开,8.5秒关,总开时间约10分钟,总处理时间约66.7分钟。
注意:超声破碎参数可能因设备而异;目标是获得均匀的裂解液(外观更白)。在4℃下以16,000× g离心30分钟,并将上清液转移到新的50 mL管中。
通过0.45 μm过滤器过滤上清液。
用缓冲液A(至少25 mL)平衡5 mL His-trap柱(GE)。
注意:如果使用过His-trap柱,可以通过NaOH、ddH2O和HCl的洗涤周期来恢复,然后将柱子浸没在5 mL ddH2O中。
清洗His-trap柱:
注意:这一步骤仅适用于恢复旧柱。a. 使用前,先用10 mL 0.1 M NiSO4、10 mL ddH2O进行洗涤,最后用20 mL缓冲液A洗涤。
b. 使用时:缓慢按压移液管,确保液体是逐滴过滤(而不是形成连续流动)。
c. 使用后:用10 mL ddH2O洗涤
用0.1 M EDTA、1 M NaCl洗涤10 mL
用ddH2O洗涤10 mL
用1 M NaOH洗涤10 mL
用ddH2O洗涤10 mL
用1 M HCl洗涤10 mL
用ddH2O洗涤10 mL
注意:柱子需要保存在水中。过滤以确保柱内无气泡。储存于4℃。使用FPLC协议进行纯化。
注意:必须提前准备好缓冲液!过滤后的柱子不能被水吸入,也不得接触其他溶液(因此要确保所有连接正确无误)。仪器设置:将起始缓冲液放入A1泵,将洗脱缓冲液放入B1泵,选择位置2的柱连接线路,将A1泵的连接点置于上游,B1泵的连接点置于下游。
a. 洗涤步骤:
i. 打开AKTA Explorer UNICORN软件,在手动模式中选择“执行”,选择洗涤中的泵 B,然后点击“入口”。
ii. 选择中间圆圈的位置2,并为过滤界面选择废液。
iii. 在自动停止后选择泵A进行洗涤,步骤同上。
iv. 最后,清洁整个系统流程,选择流量为5 mL/min,压力关闭,洗涤10分钟。
注意:使用所有缓冲液A和梯度选项为0%缓冲液B。
b. 在洗脱后,所有试管都需要更换为新的。从第1号开始,大约四十个1.5 mL的试管按顺序放置。提起出流尖端,直接移至第一个试管,并将柱子定位。
c. 在手动选项中设置流量和通道。
i. 流量:1.5 mL/min
ii. 压力:系统压力
iii. 梯度:目标4%缓冲液B
iv. 分部收集-分部体积:5 mL
v. 报警:报警系统压力-高报警1.00 Mpa
注意:设置完成后,再次检查洗脱系统是否能够自动按顺序运行。d. 一旦mAU值稳定,将梯度目标更改为10%缓冲液B,并将时间设置为15分钟,以洗涤可能非特异性结合的蛋白。这个梯度大约需要15分钟从4%缓冲液B增加到10%缓冲液B。
再次等待,直到稳定并且峰消失;然后将梯度更改为20%缓冲液B,并将时间设置为15分钟。
注意:Cas9蛋白的安全咪唑浓度范围在50到250 mM之间。达到250 mM后,如果没有峰出现,则表示杂质大量不存在。在咪唑浓度大约在50–250 mM范围内的峰处收集洗脱液。进行SDS-PAGE电泳以验证蛋白质分子量
a. 在峰流出物中,从每个试管中取8 μL的粗蛋白产物,加入2 μL的SDS-PAGE蛋白上样缓冲液(5×),并在95℃下变性10分钟。
注意:第一次流过物作为对照。标记:加入5 μL BeyoColor Prestained Color Protein marker 10–170 kDa。运行条件:150 V,30分钟。
b. 完成后,在蛋白质染色溶液中进行染色。
注意:微波加热15秒或摇动5分钟。
c. 然后,摇动30分钟。
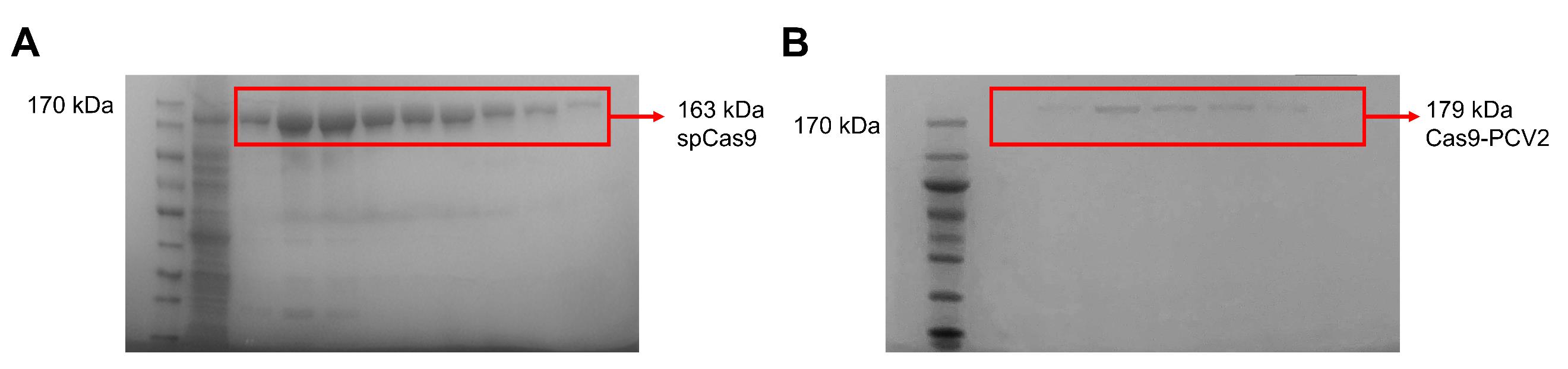
d. 检查蛋白质大小:Cas9约为163 kDa,Cas9-PCV2约为179 kDa。凝胶图像展示在图2中。e. 比较结果,找出5-6个最亮的分部,然后按照以下步骤进行蛋白质浓度测定。
注意:在运行过程中,台式离心机需预先在4℃下冷却。Fig2如下:体外纯化及活性检测Cas9和Cas9-PCV2融合蛋白。(A) 通过SDS-PAGE验证纯化的spCas9蛋白。(B) 通过SDS-PAGE验证纯化的Cas9-PCV2融合蛋白。
蛋白质浓度测定:
a. 将离心过滤器(预先在冰上冷却10 K 50 mL和3 K 15 mL过滤器)。
b. 将选定的5-6个样品分部添加到预先冷却的10 K 50 mL过滤器中。
c. 在预先冷却的离心机中离心30分钟,观察上清液,并在剩余体积约为2.5 mL时随时停止离心。使用PD-10柱对浓缩样品进行脱盐
a. 大约需要10管100 μL的考马斯亮蓝以实时检测洗脱物中是否有蛋白质存在,并比较蛋白质浓度。
b. 用Cas9储存缓冲液洗涤PD-10柱三次,每次5分钟。
c. 然后,将2.5 mL的浓缩溶液加入PD10中,随后进行离心。加入500 μL的储存缓冲液,用1.5 mL离心管收集洗脱物,每次加入500 μL,总共加入七次,收集每个500 μL产品的七个试管。
d. 取出3 μL的产品,与考马斯亮蓝混合,区分蓝色的亮度,选择最亮的五个产品管,转移到已用Cas9缓冲液交换的第二个PD-10中,进行第二次脱盐。
e. 测量亮度,挑选蓝色染色最深的四个试管,继续进行下一个蛋白质浓度的测定。
注意:在冷室中进行这些步骤;向PD-10柱中装载的体积应小于或等于2.5 mL。浓缩脱盐后的蛋白质
a. 将2 mL脱盐后的产品加入预先在4℃下冷却的3 K 15 mL离心过滤器中,然后在4,000× g的预先冷却的离心机中离心大约30分钟。注意剩余体积,并在剩余体积达到500 μL时停止离心。
b. 在冷室中,将20 μL分装到1.5 mL试管中。
c. 储存在-80℃冰箱中。
d. 保留一管产品用于浓度测定。
e. 使用NanoDrop 2000测量蛋白质在280 nm处的吸收峰。
f. 使用Expasy和AAT Bioquest软件计算最终浓度:蛋白质浓度计算器。
进行体外酶活性测试。
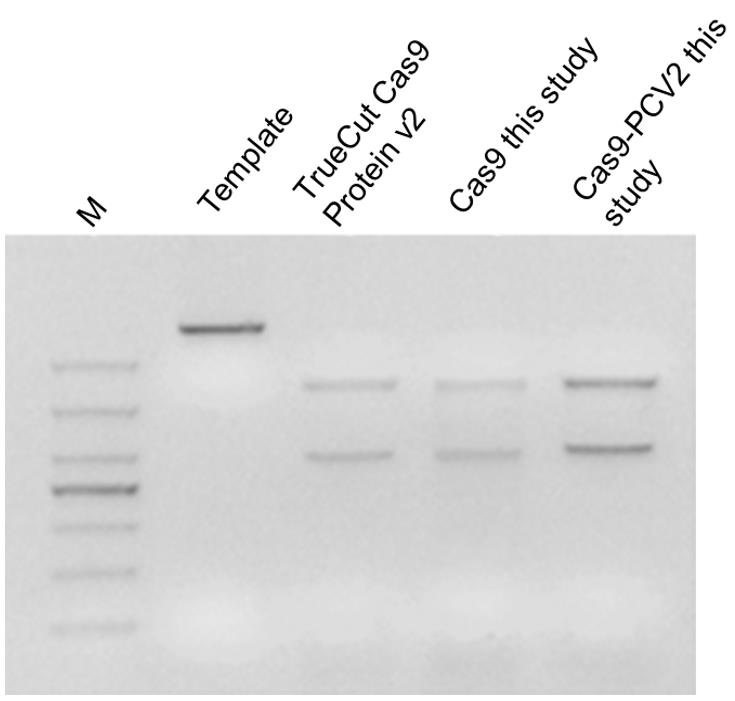
a. 通过以下体外切割实验检测Cas9蛋白的活性。
i. 在20 μL体积中建立以下切割反应。试剂和体积如表格6所示;按给定的顺序加入反应 组分。
Table 6. Reaction mixture for in vitro cleavage assayReagent Volume Cleavage buffer (10×) 2 μL Substrate DNA 100 ng Cas9 (or Cas9-PCV2) 0.3 μg sgRNA 130 ng Nuclease-free water up to 20 μL
ii. 在37°C下孵育30分钟。
iii.使用1× TAE缓冲液的3%琼脂糖凝胶电泳可视化切割带。凝胶图像展示在图3中(如下)。
b.测量Cas9-PCV2融合蛋白的酶活性。
i.由Sangon Biotech合成的ssDNA探针:PCV2-linker序列(5′→3′):BHQ1-TAAGTATTACCAGAAA/i6FAMdT/cctcttgtcccacagat atccagaaccctgaccctgccgtgtaccagct
ii.分别用ddH2O稀释PCV2连接物至最终浓度为0.02、0.06和0.10 μM。
iii.将ssDNA探针与Cas9-PCV2在37°C下共同孵育,并使用荧光分光光度计F-7000检测0.02 μM Cas9-PCV2蛋白与不同浓度的ssDNA探针共同作用时的荧光素强度。
注意:当ssDNA探针被Cas9-PCV2融合蛋白切割时,FAM荧光素从猝灭基团BHQ1中释放出来并开始发射荧光,这由荧光光谱仪实时监测。
C. 合成并纯化针对HEK293T细胞中gDNA的sgRNA(GeneArt Precision gRNA合成试剂盒)
设计如下序列,用于合成sgRNA模板组装所需的Target F1正向和Target R1反向引物。序列展示在表格7中:
Target F1:TAATACGACTCACTATAG + 目标序列(20 nt)
Target R1:TTCTAGCTCTAAAAC + 目标序列的反向互补序列(20 nt)
注意:如果目标序列中已经包含一个5′ G,您可以保留它,这将导致T7启动子引物中额外添加一个G。或者,您可以从目标序列中移除第一个G,该G将由T7启动子引物添加回来。

引物由Sangon Biotech合成,并使用无核酸酶的水稀释至最终0.3 μM浓度(工作溶液).
使用GeneArt Precision gRNA合成试剂盒进行体外sgRNA转录:
a. 在25 μL体积中建立以下PCR组装反应,并按照以下步骤准备反应组分;试剂和体积如表格8所示。
Table 8. Components to make gRNA DNA template
| Reagent | Volume |
|---|---|
| Phusion High-Fidelity PCR Master Mix (2×) | 12.5 μL |
| Tracr Fragment + T7 Primer Mix | 1 μL |
| 0.3 μM Target F1/R1 primer mix | 1 μL |
| Nuclease-free water | 10.5 μL |
b.使用以下循环参数进行组装PCR,如下表9所示。
Table 9. PCR parameters to generate gRNA DNA template
| Cycle step | Temperature | Time | Cycles |
|---|---|---|---|
| Initial denaturation | 98 °C | 10 s | 1× |
| Denaturation | 98 °C | 5 s | 32× |
| Annealing | 55 °C | 15 s | |
| Final extension | 72 °C | 1 min | 1× |
| Hold | 4 °C | Hold | 1× |
c. 进行gRNAs的体外转录(IVT);试剂和体积如表格10所示, 彻底混合试管中的内容物, 并快速离心。在37°C下设置IVT反应进行3小时。
注意:所有操作最好在RNase-free工作台中完成。
Table 10. Components of IVT reaction
| Reagent | Volume |
|---|---|
| NTP mix (100 mM each of ATP, GTP, CTP, UTP) | 8 μL |
| gRNA DNA template (from PCR assembly) | 6 μL |
| 5× TranscriptAidTM Reaction Buffer | 4 μL |
| TranscriptAidTM Enzyme Mix | 2 μL |
通过DNase I消化去除DNA模板:
在IVT反应后立即将IVT反应混合物与1 μL的DNase I(1 U/μL)一起孵育,并在37°C下孵育15分钟。
使用GeneArt gRNA清洁试剂盒纯化sgRNA。
a. 将IVT反应稀释至200 μL的无核酸酶的水中,并加入100 μL的结合缓冲液。通过抽吸混合。
b. 加入300 μL的乙醇(> 96%)并通过抽吸混合。
c. 将混合物转移到预先装有收集管的GeneJET RNA纯化微柱中,并在14,000× g下离心30–60秒。丢弃流过物。
d. 用700 μL的洗涤缓冲液1和洗涤缓冲液2洗涤结合的RNA。
e. 在14,000× g下额外离心空纯化柱60秒,以完全去除任何残留的洗涤缓冲液。
f. 将纯化柱转移到新的1.5 mL RNase-free EP管中。
g. 在柱过滤器的中心加入10 μL的无核酸酶的水,并在14,000× g下离心60秒以洗脱sgRNA。
h. 使用NanoDrop 2000测量sgRNA的浓度。
i. 将sgRNA储存在-80°C。
注意:所有操作最好在RNase-free工作台中完成。
D. 准备RNP复合物,用于odsDNA附着到Cas9-PCV2融合蛋白上
设计和合成PCV2连接物。
a. 由于PCV2蛋白识别5′至3′方向的DNA序列(AAGTATT^ACCAGAAA),odsDNA具有3′突出端,因此需要一个PCV2连接物作为桥梁来将odsDNA附着到Cas9-PCV2融合蛋白上。
注意:“^”代表PCV2蛋白的切割位置。b. PCV2连接物分为两部分:5′端是一个固定序列,被PCV2蛋白识别以进行切割,随后与PCV2蛋白形成共价连接,3′端是与odsDNA的3′突出端互补配对的序列。
c. 用于Lamin A/C位点的PCV2连接物序列如表格11所示。
Table 11. PCV2 linker sequence for the *Lamin A/C* locus
| *Lamin A/C* locus | Cas9-PCV2 linker sequence(5′→3′) |
|---|---|
| PCV2 linker | AAGTATTACCAGAAAtgcttttttt |
注意:大写字母是PCV2蛋白识别的预设序列,而小写字母是与Lamin A/C位点的odsDNA互补配对的序列。
d. PCV2连接物的合成由Sangon Biotech完成,并使用无核酸酶的水稀释至10 μM
PCV2连接物与odsDNA的退火。
a. 我们使用具有10个碱基突出端的odsDNA与具有10个配对碱基的PCV2连接物进行退火。
b. 在10 μL体积中建立以下退火反应;试剂和体积如表格12所示。
Table 12. Components of annealing reaction
| Reagent | Molar amount | Volume |
|---|---|---|
| PCV2 linker | 7.5 pmol | 0.75 μL |
| odsDNA (1 μg/μL) | 7.5 pmol | 5 μL |
| 5× annealing buffer | - | 2 μL |
| Nuclease-free water | - | to 10 μL |
c. 使用如下表13所示的循环参数进行退火反应。
Table 13. Annealing reaction to generate odsDNA/PCV2 linker complex
| Cycle step | Temperature | Time | Cycles |
|---|---|---|---|
| Denaturation | 65 °C | 5 min | 1× |
| Annealing | 65 °C (-0.1 ℃/8 s) to 25 °C | ~60 min | 1× |
| Hold | 4 °C | Hold | 1× |
准备由odsDNA/PCV2连接物/Cas9-PCV2融合蛋白组成的RNP复合物。
a. 为了验证odsDNA/PCV2连接物能否有效地附着到Cas9-PCV2蛋白上,将不同摩尔比率的0:1、1:1、2:1、3:1和4:1与Cas9-PCV2蛋白一起孵育,然后通过2%琼脂糖凝胶进行验证。如图4所示,当Cas9-PCV2融合蛋白与odsDNA/PCV2连接物的摩尔比率为3:1时,下方的游离odsDNA/PCV2连接物几乎完全反应,所有odsDNA/PCV2连接物都附着到Cas9-PCV2融合蛋白上。
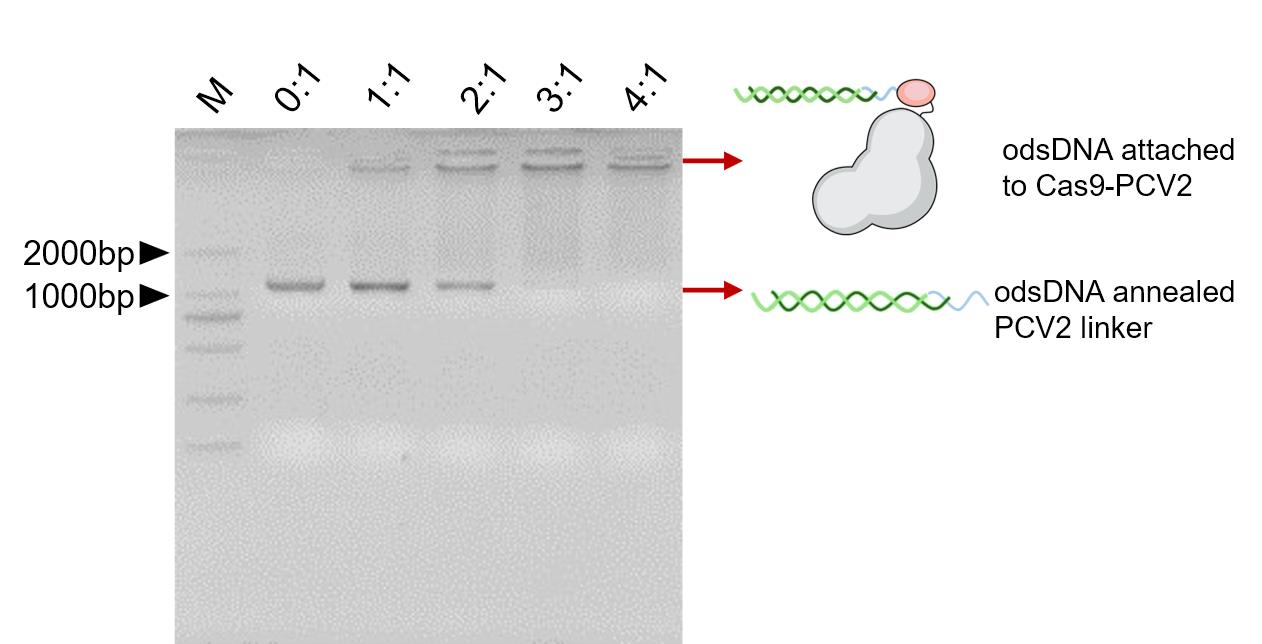
[^图4. 验证odsDNA附着到Cas9-PCV2蛋白上的情况。]: 当Cas9-PCV2与预退火的odsDNA/PCV2连接物的比例达到> 3:1时,odsDNA供体有效地连接到Cas9-PCV2上。
b. 将30 pmol的Cas9-PCV2融合蛋白添加到来自步骤D2的odsDNA/PCV2连接物复合物中。
c. 因为Cas9-PCV2融合蛋白的数量远多于odsDNA,几乎所有的odsDNA都完全反应并附着到Cas9-PCV2融合蛋白上。
d. PCV2蛋白的工作条件要求至少有1 mM的Mg2+存在。由于Cas9-PCV2蛋白的缓冲液C(储存缓冲液)中含有10 mM的MgCl2,因此这里不需要额外添加Mg2+。
E. 将odsDNA/Cas9-PCV2/sgRNA RNP复合物转染入HEK293T细胞
在进行核转染之前,至少在核转染实验前三天,使用Dulbecco改良Eagle培养基和10%胎牛血清对HEK293T细胞进行亚培养。
对HEK293T细胞进行odsDNA/Cas9-PCV2/sgRNA的核转染:
a. 细胞准备
i. 核转染的最佳细胞密度:80%–90%。更高的细胞密度可能导致核转染效率降低。
ii. 从培养细胞中移除培养基,并用1× PBS洗涤细胞一次。用至少与培养基等体积的1× PBS洗涤。
iii. 使用TrypLE Express试剂在37°C下孵育细胞约5分钟以收获细胞。
iv. 用含有10% FBS的培养基中和胰蛋白酶反应。大多数细胞(>90%)将脱离。打开并密封培养基、PBS和TrypLE,使用PARAFILM密封膜。b. 准备odsDNA/Cas9-PCV2/sgRNA RNP复合物
i. 为制备Cas9-PCV2 RNPs复合物,将odsDNA/PCV2连接物/Cas9-PCV2融合蛋白复合物与90 pmol sgRNA混合,并在37°C下孵育10分钟。
ii. 立即按照以下步骤进行核转染。
c. 核转染
i. 确保将整个补充剂添加到Nucleofector溶液中。Nucleofector溶液与补充剂的比例为4.5:1。对于单个反应,使用82 μL的Nucleofector溶液加上18 μL的补充剂,总共100 μL的总体积。
ii. 通过填充适当的孔数,用1 mL补充的培养基预热/平衡6孔板,并在含5% CO2的湿润条件下,在37°C下预热/平衡板。
iii. 通过胰蛋白酶消化收获细胞。
iv. 使用血细胞计数器或自动细胞计数器(视个人喜好)计数细胞数量。
v. 在室温下,以200× g离心10分钟所需的细胞数(每个样本1 × 106细胞)。完全去除上清液。
vi. 小心地将细胞沉淀在每样本100 μL室温Nucleofector溶液中重新悬浮。
注意:不要让细胞在Nucleofector溶液中停留过长时间(即超过15分钟),因为这可能会降低细胞存活率和基因转移效率。
i. 将100 μL的细胞悬液与odsDNA/Cas9-PCV2/sgRNA复合物结合。表格14显示了不同的样本。
Table 14. One Nucleofection® sample contains
| odsDNA sample | odsDNA sample | dsDNA sample |
|---|---|---|
| 1 × 10^6 HEK293T cells | 1 × 10^6 HEK293T cells | 1 × 10^6 HEK293T cells |
| odsDNA/Cas9-PCV2/sgRNA complex | odsDNA, Cas9/sgRNA complex | dsDNA, Cas9/sgRNA complex |
| 100 μL Cell Line Nucleofector® Solution V | 100 μL Cell Line Nucleofector® Solution V | 100 μL Cell Line Nucleofector® Solution V |
ii. 将细胞/DNA悬液转移到一个认证过的细胞培养皿(样本必须覆盖皿底而没有气泡)。用盖子关闭培养皿。
iii. 为Nucleofector® 2b设备选择合适的Nucleofector®程序D-032。
iv. 将含有细胞/DNA悬液的培养皿插入Nucleofector®培养皿夹持器中,并通过按下X按钮应用所选程序。
v. 一旦程序完成,从夹持器中取出培养皿。
vi. 立即用约500 μL的预平衡培养基补充培养皿,并轻轻将混合物转移到已准备好的6孔板中(每个孔的最终体积为1.5 mL培养基)。使用提供的移液器,并避免对样本进行重复抽吸。
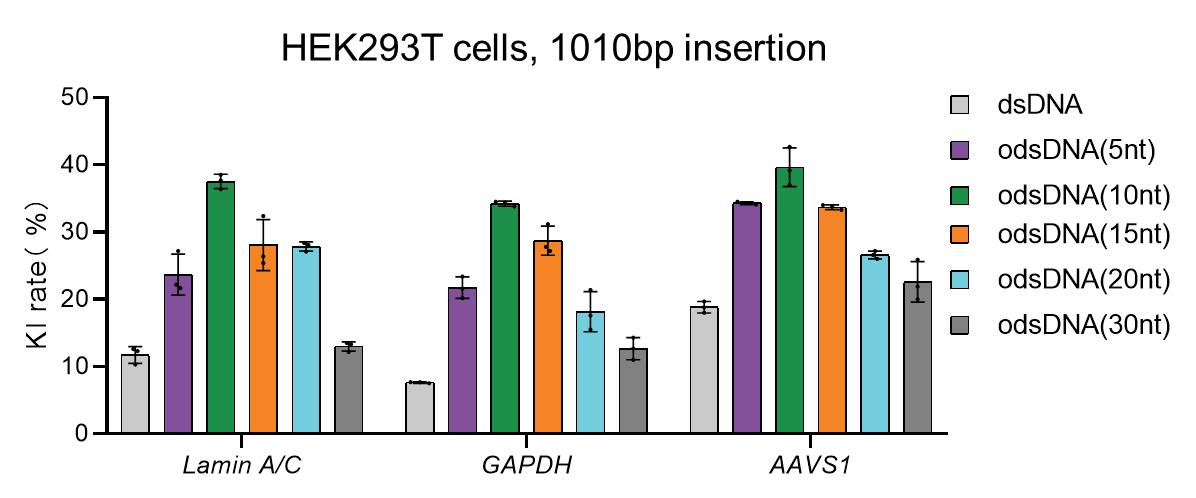
[^图5. 使用不同供体在HEK293T细胞中进行敲入(KI)的效率。]: 选择了三个不同的基因组位点用于1,010 bp odsDNA供体的KI,dsDNA供体作为对照。数据在核转染后15天收集。KI效率是根据总EGFP阳性细胞数(供体KI)除以总RNP电转染细胞数计算得出的平均百分比。
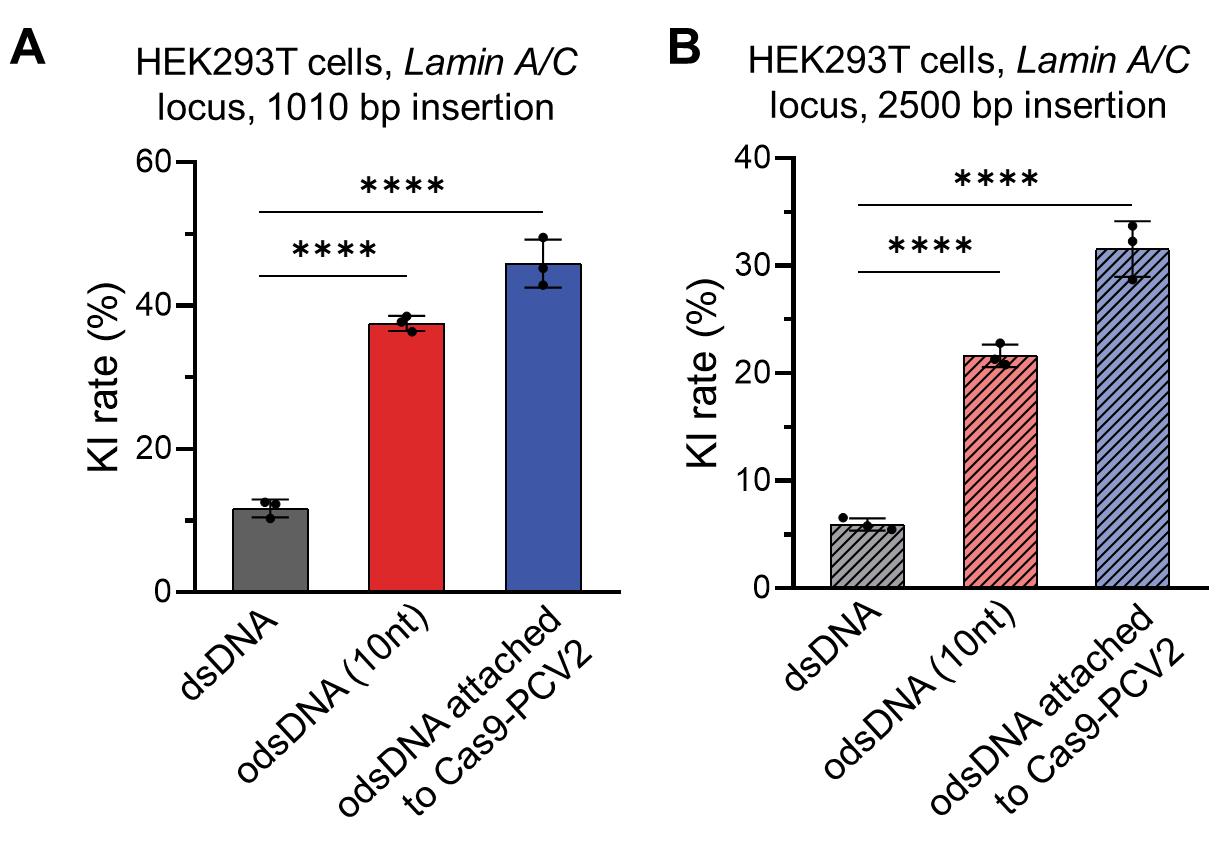
[^图6. 使用odsDNA附着到Cas9-PCV2蛋白策略进行不同长度的DNA片段敲入(KI)的比较。]: (A) 1,010 bp的供体模板,包括Cas9结合的双链DNA和10 nt的单链DNA,以及Cas9-PCV2结合的10 nt单链DNA,被电转染到HEK293T细胞以靶向Lamin A/C位点。KI效率被绘制在一起进行比较。数据在核转染后15天收集。(B) 2,500 bp的供体模板,包括Cas9结合的双链DNA和10 nt的单链DNA,以及Cas9-PCV2结合的10 nt单链DNA,被电转染到HEK293T细胞以靶向Lamin A/C位点。KI效率被绘制在一起进行比较。数据在核转染后21天收集。
材料、试剂和配方:
配方:
LB medium (1,000 mL)
Reagent Final concentration Quantity TryptoneYeast extractNaCl 10 g/L5 g/L10 g/L 10 g5 g10 g ddH2O n/a ~980 mL Total n/a 1,000 mL
调整pH值至7.4,使用氢氧化钠。在121°C下高压灭菌15分钟。在室温下储存。
- 50 mg/mL kanamycin (10 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| Kanamycin | 50 mg/mL | 0.5 g |
| ddH2O | n/a | 10 mL |
| Total | n/a | 10 mL |
使用0.22 μm注射器过滤器对溶液进行无菌过滤。在4°C下储存。
- LB agar plate with kanamycin (1,000 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| TryptoneYeast extractNaCl | 10 g/L5 g/L10 g/L | 10 g5 g10 g |
| Agar | 15 g/L | 15 g |
| 50 mg/mL Kanamycin | 50 μg/mL | 1 mL |
| ddH2O | n/a | ~960 mL |
| Total | n/a | 1,000 mL |
a. 调整pH值至7.4,使用氢氧化钠。
b. 在121°C下高压灭菌15分钟。灭菌后加入琼脂。
c. 在搅拌板上搅拌培养基,并允许其冷却至30–40°C。
d. 加入1 mL 50 mg/mL卡那霉素溶液。充分搅拌。
e. 用无菌移液管将25 mL溶液转移到10 cm的培养皿中。
f. 在层流罩中让琼脂平板冷却并干燥30分钟。
g. 将平板放入塑料袋中,在4°C下储存。
- Terrific broth medium (1,000 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| Terrific brothGlycerol | 47.6 g/L0.4% (v/v) | 47.6 g4 mL |
| ddH2O | n/a | ~960 mL |
| Total | n/a | 1,000 mL |
调整pH值至大约7.4,使用氢氧化钠。在121°C下高压灭菌15分钟。在室温下储存。
- 1 M IPTG (10 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| IPTG | 1 M | 2.38 g |
| ddH2O | n/a | ~9.8 mL |
| Total | n/a | 10 mL |
使用0.22 μm注射器过滤器对溶液进行无菌过滤。在-20°C下储存。
- Buffer A: start buffer (1,000 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| TrisNaCl | 20 mM500 mM | 2.4228 g29.22 g |
| ddH2O | n/a | ~960 mL |
| Total | n/a | 1,000 mL |
调整pH值至大约8.0,使用盐酸。使用Nalgene Rapid-Flow对溶液进行无菌过滤。在4°C下储存。
- Buffer B: elution buffer (1,000 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| TrisNaCl | 20 mM500 mM | 2.4228 g29.22 g |
| imidazole | 500 mM | 34.04 g |
| ddH2O | n/a | ~960 mL |
| Total | n/a | 1,000 mL |
调整pH值至大约8.0,使用盐酸。使用Nalgene Rapid-Flow对溶液进行无菌过滤。在4°C下储存。
- Buffer C: storage buffer (1,000 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| TrisKCl | 20 mM200 mM | 2.4228 g14.91 g |
| MgCl2·6H2O | 10 mM | 2.03 g |
| Glycerol | 10% (v/v) | 100 mL |
| ddH2O | n/a | ~860 mL |
| Total | n/a | 1,000 mL |
调整pH值至大约8.0,使用盐酸。使用Nalgene Rapid-Flow对溶液进行无菌过滤。在4°C下储存。
- Buffer D: cleavage buffer (1,000 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| HEPESKCl | 20 mM150 mM | 4.77 g11.18 g |
| MgCl2·6H2O | 1 mM | 0.2 g |
| TCEP | 1 mM | 0.25 g |
| Glycerol | 10% (v/v) | 100 mL |
| ddH2O | n/a | ~860 mL |
| Total | n/a | 1,000 mL |
调整pH值至大约8.0,使用盐酸。使用Nalgene Rapid-Flow对溶液进行无菌过滤。在4°C下储存。
- 0.1 M NiSO4 (100 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| NiSO4·6H2O | 0.1 M | 2.63 g |
| ddH2O | n/a | ~96 mL |
| Total | n/a | 100 mL |
使用0.22 μm注射器过滤器对溶液进行无菌过滤。在室温下储存。
- 0.1 M EDTA, 1M NaCl (100 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| EDTA | 0.5 M | 2.92 g |
| NaCl | 1 M | 5.84 g |
| ddH2O | n/a | ~96 mL |
| Total | n/a | 100 mL |
调整pH值至8.0,使用氢氧化钠。使用0.22 μm注射器过滤器对溶液进行无菌过滤。在室温下储存。
- 1 M NaOH (100 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| NaOH | 1 M | 4 g |
| ddH2O | n/a | ~96 mL |
| Total | n/a | 100 mL |
室温存放。
- 1 M HCl (120 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| 12 M HCl | 1 M | 10 mL |
| ddH2O | n/a | 110 mL |
| Total | n/a | 120 mL |
室温存放。
- SDS-PAGE running buffer (1,000 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| TrisGlycine | 125 mM1.25 M | 15.1 g94 g |
| SDS | 0.5% (w/v) | 5 g |
| ddH2O | n/a | ~800 mL |
| Total | n/a | 1,000 mL |
室温存放。
- Low-EDTA TE buffer, 1× (for DNA) (100 mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| TrisEDTA | 10 mM0.2 mM | 0.1211 g0.0012 g |
| ddH2O | n/a | ~100 mL |
| Total | n/a | 100 mL |
调整pH值至8.0,使用盐酸。使用0.22 μm注射器过滤器对溶液进行无菌过滤。在室温下储存。
试剂:
ddH2O,DEPC处理水(Sangon Biotech,目录号:B300592)
Lambda外切酶(New England Biolabs,目录号:M0262L)
GeneJET PCR纯化试剂盒(Thermo Fisher Scientific,目录号:K0702)
SpCas9表达质粒(Addgene,目录号:47327)
Dulbecco改良Eagle培养基(DMEM)(Gibco,目录号:C11885500BT)
TrypLE Express(Gibco,目录号:12604039)
DPBS(无Ca2+或Mg2+)(Gibco,目录号:14190-144)
胎牛血清(FBS)(Gibco,目录号:10091148)
细胞系核转染试剂盒V(Lonza,目录号:VCA-1003)
Q5高保真2×主混合液(New England Biolabs,目录号:M0492L)
50× TAE缓冲液(Sangon Biotech,目录号:B548101)
6×凝胶上样染液紫色(New England Biolabs,目录号:B7024S)
SDS-PAGE蛋白质上样缓冲液,5×(Beyotime,目录号:P0286)
BeyoColor预染彩色蛋白质分子量标准,10–170 kDa(Beyotime,目录号:P0077)
Imperial蛋白质染色溶液(Thermo Fisher Scientific,目录号:24615)
DNA寡核苷酸退火缓冲液,5×(Beyotime,目录号:D0251)
LB培养基(见配方)
50 mg/mL卡那霉素(见配方)
含卡那霉素的LB琼脂平板(见配方)
Terrific肉汤(TB)培养基(见配方)
1 M IPTG(异丙基硫代-β-D-1-半乳糖吡喃糖苷)(见配方)
缓冲液A:起始缓冲液(见配方)
缓冲液B:洗脱缓冲液(见配方)
缓冲液C:储存缓冲液(见配方)
缓冲液D:切割缓冲液(见配方)
0.1 M NiSO4(硫酸镍)(见配方)
0.1 M EDTA,1 M NaCl(乙二胺四乙酸,氯化钠)(见配方)
1 M NaOH(氢氧化钠)(见配方)
1 M HCl(盐酸)(见配方)
SDS-PAGE运行缓冲液(见配方)
低EDTA TE缓冲液(见配方)
材料:
移液器吸头:10、200 和 1,000 μL(Yeasen,目录号:83010ES20、83040ES20 和 83070ES08)
200 μL延长型移液器吸头,已灭菌(Yeasen,目录号:83061ES50)
移液器:2.5、10、20、100、200 和 1,000 μL(Eppendorf,目录号:3120000216、3120000224、3120000232、3120000240、3120000259 和 3120000267)
0.2 mL PCR八联管(Yeasen,目录号:83602ES10)
1.5 mL微型离心管(Axygen,目录号:MCT-150-C)
50 mL锥形管(Thermo Fisher,目录号:339652)
DNA寡核苷酸引物和PCV2连接器(Sangon Biotech)
6孔细胞培养板(BIOFIL,目录号:TCP011006)
60 mm细胞培养皿(BIOFIL,目录号:TCD010060)
10 cm细胞培养皿(BIOFIL,目录号:TCD010100)
HEK293T细胞(美国型培养物收藏所,目录号:CRL-3216)
DH5α感受态细胞(Sangon Biotech,目录号:B528413)
大肠杆菌Rosetta 2 (DE3)株(Millipore EMD,目录号:71397)
异丙基硫代-β-D-1-半乳糖吡喃糖苷(IPTG)(Invitrogen,目录号:15529019)
胰蛋白胨(Gibco,目录号:211705)
酵母提取物(Gibco,目录号:211931)
硫酸卡那霉素(Sangon Biotech,目录号:A600286)
琼脂(Thermo Fisher Scientific,目录号:22700025)
Terrific肉汤(Thermo Fisher Scientific,目录号:22711022)
甘氨酸(Invitrogen,目录号:15527013)
SDS(Sigma-Aldrich,目录号:436143)
氯化钠(Sigma-Aldrich,目录号:S9888)
氯化钾(Sigma-Aldrich,目录号:P3911)
六水合氯化镁(Sigma-Aldrich,目录号:M2670)
三羟甲基氨基甲烷(Invitrogen,目录号:A32355)
盐酸(HCl)(Sigma-Aldrich,目录号:30721)
氢氧化钠(NaOH)(Sigma-Aldrich,目录号:221465)
咪唑(Imidazole)(Sigma-Aldrich,目录号:I2399)
UltraPure甘氨酸(Invitrogen,目录号:15527013)
β-巯基乙醇(Gibco,目录号:21985023)
EDTA(乙二胺四乙酸)(Thermo Fisher Scientific,目录号:17892)
HEPES(Sigma-Aldrich,目录号:54457)
TCEP(三(2-羧乙基)膦酸)(Thermo Fisher Scientific,目录号:T2556)
甘油(Glycerol)(Thermo Fisher Scientific,目录号:17904)
HisTrap快速流动,5 mL(GE Healthcare,目录号:GE17-5255-01)
Amicon Ultracel-100再生纤维素膜,50 mL(Millipore,目录号:UFC910008)
Amicon Ultracel-30再生纤维素膜,4 mL(Millipore,目录号:UFC803008)
0.22 μm注射器过滤器,PES膜,33 mm直径(Millipore,目录号:SLGPR33RS)
0.45 μm注射器过滤器,PES膜,33 mm直径(Millipore,目录号:SLHPR33RS)
Nalgene Rapid-Flow带PES 500 mL(Thermo Fisher Scientific,目录号:566-0020)
PD-10脱盐柱(GE Healthcare,目录号:GE17-0851-01)
BeyoGel SDS-PAGE预制凝胶,Tris-Gly,4%–20%,12孔(Beyotime,目录号:P0057A)
琼脂糖(Agarose)(Thermo Fisher Scientific,目录号:R0491)
GeneArt Precision gRNA合成试剂盒(Invitrogen,目录号:A29377)
Invitrogen TrueCut Cas9蛋白 v2(Thermo Fisher Scientific,目录号:A36497)
PARAFILM密封膜(Sigma-Aldrich,目录号:HS234526B-1EA)
Kimwipes一次性擦拭纸(Sigma-Aldrich,目录号:Z188956)
设备
Amaxa Nucleofector IIb设备(Lonza,型号:AAB-1001)
BD Accuri C6流式细胞仪(BD Biosciences,目录号:23432)
Heracell VIOS 160i CO2铜室培养箱(Thermo Fisher Scientific,目录号:51030476)
生物安全柜(Esco Airstream,型号:AB2-4S8-CN)
超声波处理器Q125(Qsonica,型号:Q125-220)
1/4直径超声波探针(Qsonica,目录号:4435)
蛋白纯化系统(AKTA pure,目录号:29046665)
荧光分光光度计F-7000(HITACHI,型号:F-7000)
IX71倒置显微镜(Olympus,型号:IX71)
雪花制冰机(XUEKE,型号:IMS-20)
水浴锅(EYELA,型号:NTT-2200)
迷你凝胶DNA电泳系统(Thermo Fisher,型号:B1)
凝胶成像系统(Azure Biosystems,型号:C300)
Veriti 96-孔PCR热循环仪(Applied Biosystems,目录号:4375786)
TAdvanced双通道PCR热循环仪(Biometra,目录号:2070212)
NanoDrop 2000分光光度计(Thermo Fisher Scientific,型号:ND-2000)
pH计(Mettle Toledo,目录号:51302807)
Milli-Q纯水系统(Millipore,型号:MP0024)
Eppendorf离心机5424 R(Eppendorf,目录号:5404000090)
Eppendorf高速冷冻离心机5910 R(Eppendorf,目录号:5942000598)
Thermo振荡器(Yeasen,目录号:80440ES03)
软件
Prism v8,用于统计和数据可视化(GraphPad,版本 8.0.1.244)
Snapgene,查看DNA序列和引物设计(版本 4.2.4)
NUPACK:核酸包装(https://nupack.org/)
Tm计算器(版本 1.16.5,https://tmcalculator.neb.com/#!/main)
CHOPCHOP,一个基于网络的sgRNA设计工具。该网站仅供非盈利和学术使用(https://chopchop.cbu.uib.no/)
Expasy,将DNA序列转换为氨基酸序列(https://web.expasy.org/translate/)
AAT Bioquest:蛋白质浓度计算器(https://www.aatbio.com/tools/calculate-protein-concentration)
NEBioCalculator(https://nebiocalculator.neb.com/#!/dsdnaamt)
该协议或其部分内容已在以下研究文章中使用并验证:
- Han et al. (2023). Efficient precise integration of large DNA sequences with 3′-overhang dsDNA donors using CRISPR/Cas9. Proc. Natl. Acad. Sci. U.S.A. (Figure 4, panel B, C and D).
- Tei et al. (2023). Comparable analysis of multiple DNA double-strand break repair pathways in CRISPR-mediated endogenous tagging. bioRxiv. (Figure 3, panel C and D).
References
- Cruz-Becerra, G. and Kadonaga, J. T. (2020). Enhancement of homology-directed repair with chromatin donor templates in cells. eLife 9: e55780.
- Doudna, J. A. (2020). The promise and challenge of therapeutic genome editing. Nature 578(7794): 229–236.
- Gu, B., Posfai, E. and Rossant, J. (2018). Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 36(7): 632–637.
- Han, W., Li, Z., Guo, Y., He, K., Li, W., Xu, C., Ge, L., He, M., Yin, X., Zhou, J., et al. (2023). Efficient precise integration of large DNA sequences with 3′-overhang dsDNA donors using CRISPR/Cas9. Proc. Natl. Acad. Sci. U. S. A. 120(22): e2221127120.
- Hsu, P. D., Lander, E. S. and Zhang, F. (2014). Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 157(6): 1262–1278.
- Maruyama, T., Dougan, S. K., Truttmann, M. C., Bilate, A. M., Ingram, J. R. and Ploegh, H. L. (2015). Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33(5): 538–542.
- Quadros, R. M., Miura, H., Harms, D. W., Akatsuka, H., Sato, T., Aida, T., Redder, R., Richardson, G. P., Inagaki, Y., Sakai, D., et al. (2017). Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 18(1): e1186/s13059-017-1220-4.
- Shy, B. R., Vykunta, V. S., Ha, A., Talbot, A., Roth, T. L., Nguyen, D. N., Pfeifer, W. G., Chen, Y. Y., Blaeschke, F., Shifrut, E., et al. (2023). High-yield genome engineering in primary cells using a hybrid ssDNA repair template and small-molecule cocktails. Nat. Biotechnol. 41(4): 521–531.
- Yu, Y., Guo, Y., Tian, Q., Lan, Y., Yeh, H., Zhang, M., Tasan, I., Jain, S. and Zhao, H. (2020). An efficient gene knock-in strategy using 5′-modified double-stranded DNA donors with short homology arms. Nat. Chem. Biol. 16(4): 387–390.
- 本文作者: Anderson
- 本文链接: http://nikolahuang.github.io/2024/03/11/在哺乳动物细胞中通过具有 3突出端的长dsDNA介导的CRISPR敲入(LOCK)实现高效的大DNA片段敲入/
- 版权声明: 转载请注明出处,谢谢。