

转载

宏基因组中挖掘原核基因组的分析流程
从宿主相关的短读长鸟枪宏基因组测序数据中恢复原核基因组
Recovering prokaryotic genomes from host-associated, short-read shotgun metagenomic sequencing data
Nature Protocols [IF:10.419]
2021-04-16 Articles
DOI: https://doi.org/10.1038/s41596-021-00508-2
第一作者:Sara Saheb Kashaf1,2
通讯作者:Robert D. Finn1,2,3*(rdf@ebi.ac.uk)
其它作者:Alexandre Almeida, Julia A. Segre
主要研究单位
欧洲生物信息学研究所,英国欣克斯顿惠康基因组校区(European Bioinformatics Institute (EMBL–EBI), Wellcome Genome Campus, Hinxton, UK)
美国国立卫生院(Microbial Genomics Section, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA)
摘要
从鸟枪法宏基因组序列数据中恢复基因组可以对微生物群落中的单个物种或菌株进行详细的分类和功能表征。恢复这些宏基因组组装的基因组 (MAG) 涉及七个阶段。首先,去除低质量碱基以及接头和宿主序列。其次,组装重叠序列以创建更长的连续片段。第三,根据序列组成和丰度对这些片段进行聚类。第四,这些序列簇或箱经过多轮质量评估和提纯以产生MAG。可选的第五阶段是 MAG 的去冗余以选择代表。接下来,对每个 MAG 进行物种分类。可选的第七阶段是评估已恢复的多样性比例。该方法的输出是基因组草图,它可以提供有关未培养生物的宝贵线索。该方法需要大约 1 周的时间运行,具体取决于可用的计算资源,并且需要具有高性能计算、shell 脚本编程和 Python 的经验。
背景
从历史上看,细菌群落的调查是通过培养研究进行的,这些研究偏向于在标准实验室条件下易于培养的物种。基于 DNA 的独立于培养的革命性方法为探索微生物群落开辟了新的窗口。直接从样本中提取和测序 DNA,无需事先在培养物中富集,可以减少对细菌群落的偏见调查。最常见的基于 DNA 的方法之一是鸟枪宏基因组测序,它涉及从生物样本中分离和测序 DNA。这反过来又允许推断有关已测序微生物群落的功能和分类信息,这将根据所使用的样本收集、DNA 提取和文库构建方法而有所不同 。对鸟枪法宏基因组序列数据进行分类分析的一种方法涉及将序列读长与来自培养细菌的参考基因组进行比较。这些基于读长的方法受到参考数据库不完整的限制。进一步限制基于读长的比对方法的效用是在不同菌株之间观察到的遗传异质性 ,其中参考菌株的遗传差异导致比对读长属于其他菌株的失败。一种分析鸟枪法宏基因组序列数据的补充方法依赖于从头组装和分箱来重建宏基因组组装的基因组 (MAG)。这种方法的一个主要优点是它允许恢复尚未分离和培养生物体的基因组,因此参考数据库中不存在。此外,基因组草图的重建提供了更广泛的基因组背景,以允许将单个分类群与特定功能联系起来。然而,由于用于生成 MAG 的计算方法的性质,它们可能会受到来自其他相关生物、高度碎片化和/或不完整的基因组序列的污染。因此,使用 MAG 需要特殊的方法考虑,不应简单地将其视为分离基因组的替代品。尽管与分离基因组相比,它们的质量较低,但 MAG 重建有助于发现大量新生物和功能。
方法的开发
Development of the protocol
通过对人类肠道宏基因组数据集的大规模分析,Almeida 等人能够恢复大量新的细菌多样性。更具体地说,通过对 11,850 个人类肠道微生物组样本的分析,可以使用本方法中讨论的一些方法恢复1,952 种未培养的候选细菌物种。这些新的细菌物种在来自世界上研究不足地区的样本中特别丰富,使非洲和南美样本的读取分配改进了 200% 以上。通过结合来自公共人类肠道微生物组数据集的 200,000 多个培养的基因组和 MAG,Almeida 等人创建了统一的人类胃肠基因组 (UHGG)集。这些基因组的功能分析被用于生成统一人类胃肠蛋白 (UHGP)集,这表明新的基因组包含大量未开发的功能多样性。这两种资源可用于推动未来人类肠道微生物组的分类和功能分析。在本方法中,我们使用 Almeida 等人探索的宏基因组数据集的一个子集展示了我们的流程(图 1)。我们还简要讨论了这些方法如何应用于我们对人类皮肤微生物组的研究。我们选择了皮肤和肠道,因为它们各自代表一个具有独特特性的生态位:肠道是一个更加多样化和微生物丰富的环境,而人类皮肤样本通常具有较低的微生物生物量,宿主污染的比例更大。
图1:从鸟枪法宏基因组中恢复原核 MAG 的工作流程概述
Fig. 1: Overview of workflow for recovery of prokaryotic MAGs from shotgun metagenomic sequence data.
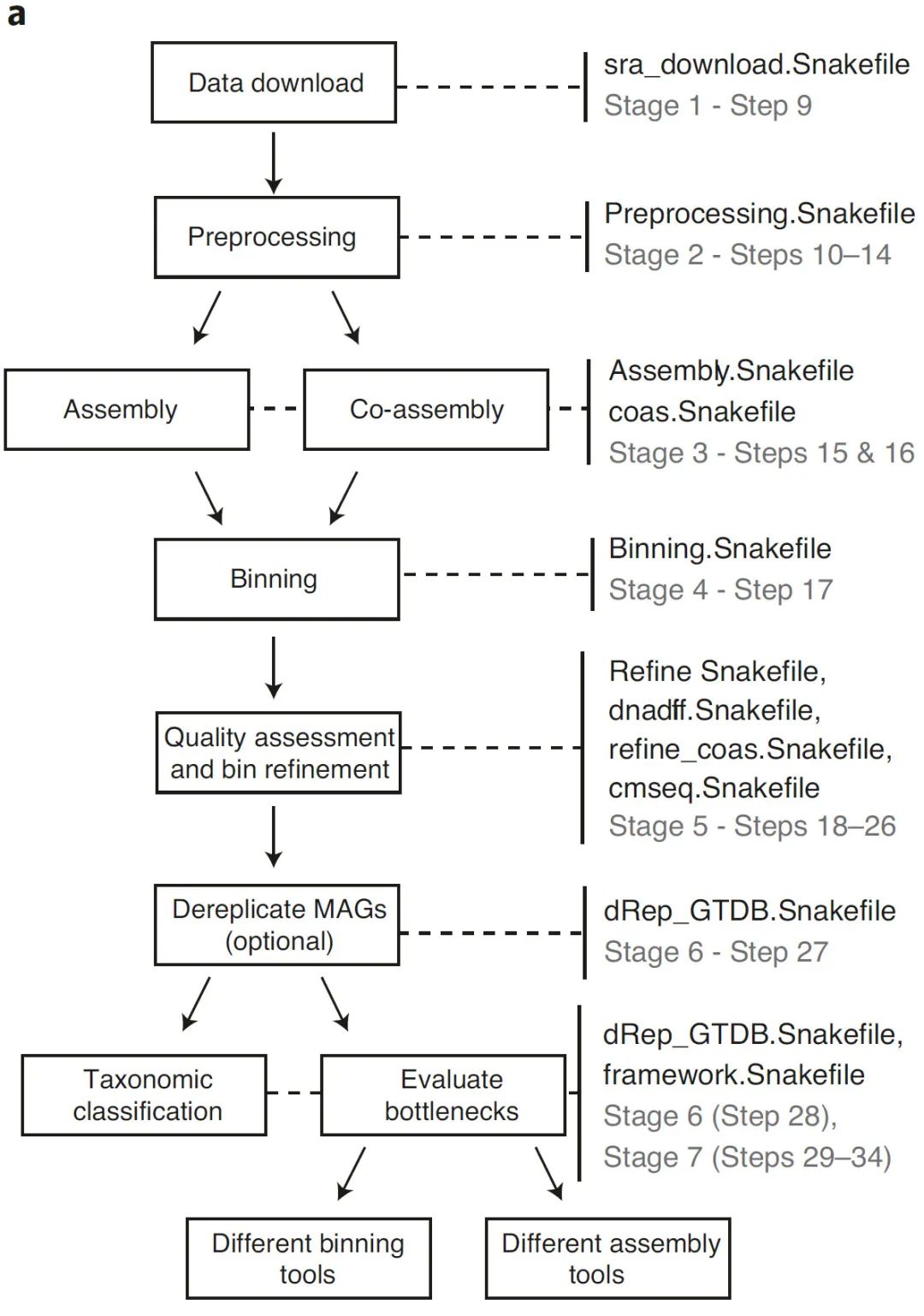
a,我们的工作流程示意图和用于执行每个步骤的 Snakemake 模块。
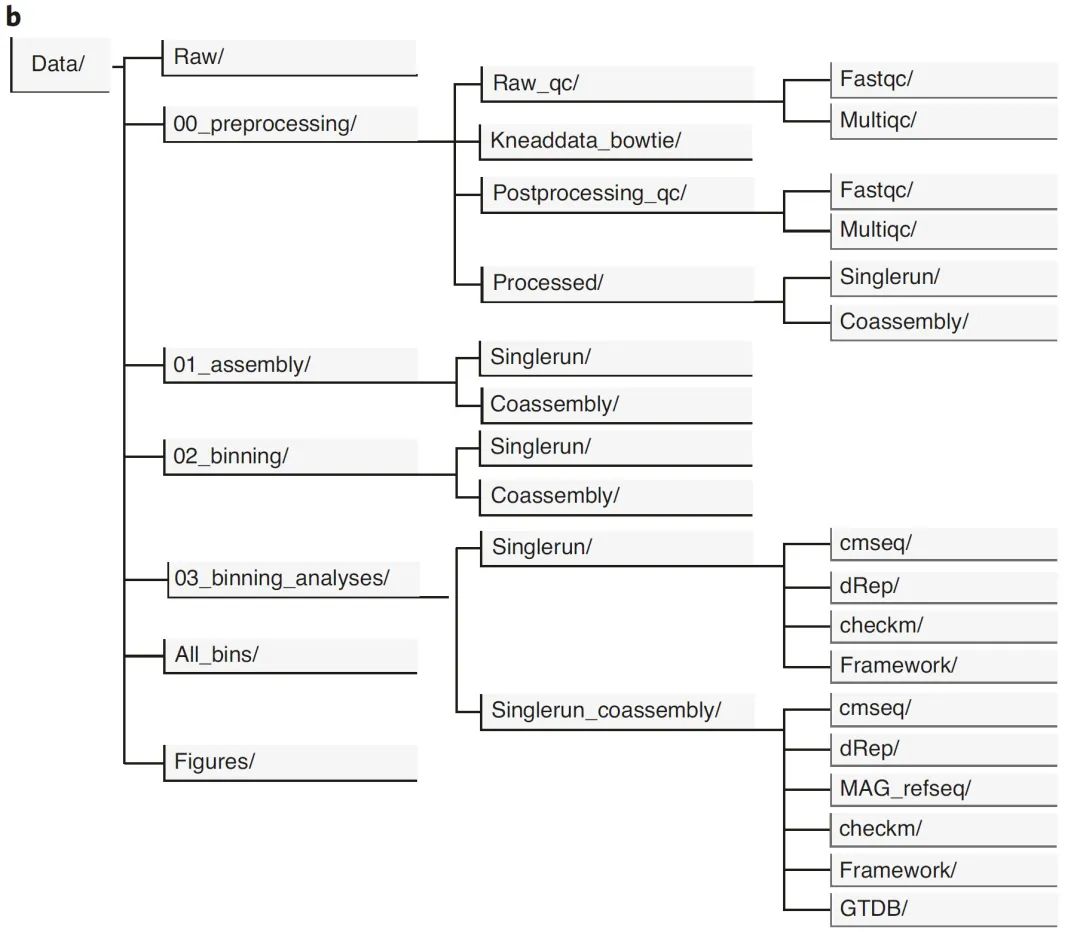
b,运行我们的 Snakemake 流程所期望的目录结构。
优点和局限性
Advantages and limitations
短读长测序技术,例如来自 Illumina 和 BGI 的技术,具有成本效益和准确度,并且具有高通量。从这些短读长测序技术生成的 MAG 已被证明可以在不同的数据集上重现,并极大地扩展了我们对生命之树的了解。然而,基于短读长测序数据集的方法在解析高度重复的基因组或其他高度保守的序列(例如在核糖体 RNA 基因中发现的序列)方面受到限制。长读长测序仪,例如 Pacific Biosciences 的 SMRT或 Oxford Nanopore 的 MinION,可以产生长度为 10 kb 或更长的读段。通过跨越这些重复区域的长度,长读长测序可以显着提高宏基因组组装的质量,但目前以降低核苷酸准确性为代价,这个问题可以通过结合短读长和长读序列数据来部分解决,称为混合组装。随着长读长测序技术在准确性和通量方面的性能不断提高,它们很可能会扩大其在宏基因组研究中的应用。
从宏基因组数据集重建基因组草图的局限性在于,由于各种技术原因(例如,样本测序深度通常不够高,无法充分覆盖存在的低丰度物种,高完整性和低污染的 MAG 可能仅代表整个宏基因组样本中一小部分)。一项研究检查了测序工作对使用计算机方法从 Illumina 序列数据集中恢复 MAG 的影响。作者发现,恢复中等和高质量 MAG 所需的最小测序阈值在 0.6 到 ~6.1 Gb 之间变化,但随着测序工作的增加,MAG 的数量开始饱和。此外,恢复 MAG 所需的覆盖深度取决于样本的多样性。一项比较不同组装程序的研究表明,×10 的覆盖率足以恢复样本中独特物种的基因组,但当同一样本中存在密切相关的菌株时,需要更高的覆盖率 (×50)。
MAG 的另一个主要限制是,与分离基因组相比,它们是从假定代表密切相关类群的重叠群的聚类中计算得出的。分箱算法有时无法区分来自不同生物体的重叠群,导致嵌合基因组草图(受污染的基因组)的恢复。他们也经常无法正确地整合来自质粒和其他移动遗传元件的重叠群。
最后,该方法的一个限制是我们的流程由许多不同的工具组成,每个工具都支持各种不同的输入和输出。本方法的重点主要是这些工具的具体应用,而不是讨论这些工具的所有可能的输入和输出。对于每个步骤,也可以有多种工具可以执行类似的角色。比较不同工具的优点不在本文的一般范围内。在故障排除部分,我们还讨论了可能出现的部分问题,但我们预计其他问题可以在与每个工具关联的 GitHub 问题部分中解决,或者如果适用,在与此方法关联的 GitHub (https://github.com/Finn-Lab/MAG_Snakemake_wf )。
实验设计
该方法的输入是一组宏基因组,在此称为测序批次。更具体地说,输入是之前由 Almeida 等人分析的人类肠道微生物组测序数据。为了展示我们的方法如何适应从其他宿主相关的生物群落中恢复多样性,我们在讨论我们方法的第七阶段时简要讨论了我们对一组人类皮肤运行的分析。该皮肤数据集由 Oh 等人 从 594 个样本生成,可从 SRA 下载,登录号为 PRJNA46333。该方法的步骤 1-9 涉及我们工作流程的设置,包括安装软件和下载必要的数据和数据库。
阶段 1:预处理(步骤 10-14)
在开始分析鸟枪法宏基因组测序数据时,需要进行彻底的质量控制,以确保原始数据具有足够的质量,并且数据不存在可能影响下游分析的偏差或问题。FastQC为数据创建质量报告,可以提供读长质量和接头序列分布的概览(图 2)。此外,根据所研究的样本,鸟枪法宏基因组测序数据可能包含高比例的宿主与微生物读长。在这种情况下,可以使用 KneadData (http://huttenhower.sph.harvard.edu/kneaddata) 等工具过滤宿主测序读长,该流程可以执行质量过滤、接头修剪和宿主净化。KneadData 使用 Trimmomatic 修剪接头序列并去除低质量读长。它还使用 Bowtie2 来丢弃与宿主基因组可比对的序列。数据预处理完成后,可以再次使用 FastQC 来可视化变化。
图 2:预处理后宏基因组序列数据集的 MULTIQC 图
Fig. 2: MULTIQC plots of metagenomic sequence datasets after preprocessing.
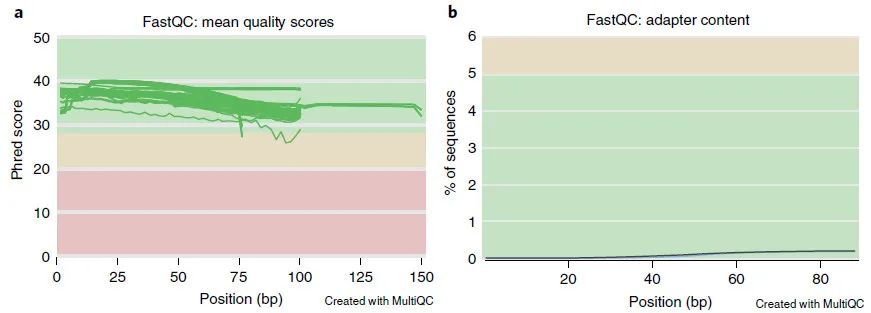
a,预处理后肠道数据集的 MULTIQC 图显示,对于每个读长文件 (FASTQ),该位置所有碱基的每个位置的平均质量(Phred 分数)。绿色、橙色和红色的阴影将 y 轴质量值分别划分为质量非常好、质量合理和质量低的得分。每条绿线代表一个不同的读长文件。该图显示,经过预处理后,读长中每个碱基位置的平均质量值具有合理或非常好的质量。b,预处理后肠道数据集的接头含量的 MULTIQC 图。每行显示不同读长文件的接头序列的百分比。接头读长的百分比随着读长长度的增加而增加,因为一旦在读取中检测到接头序列,就假定它一直存在到读长的末尾。鉴于接头序列的百分比 <5%(由绿色阴影表示),下游分析可以继续。
阶段 2:组装(步骤 15 和 16)
下一步是组装,包括将重叠的宏基因组读长合并到更长的连续 DNA 片段(重叠群)中。这些重叠群中的一些被排序并跨间隙合并以形成支架。基于德布鲁因图算法的方法通常用于鸟枪宏基因组序列数据,并且是本方法的重点。德布鲁因图组装软件的选择取决于科学问题和研究人员可用的计算资源。对组装软件的比较将这些工具分为以基因为中心的工具,例如 metaSPAdes 和 IDBA-UD,以及以基因组为中心的工具,例如 SPAdes。SPAdes 最初是为单细胞测序数据开发的,尽管其内存消耗很高,但已成功应用于一些低复杂度宏基因组的研究中。MetaSPAdes 是 SPAdes 工具包的宏基因组学流程,与 SPAdes 相比,它减少了内存使用量并改善了运行时间,因此更适合大型复杂的宏基因组数据集。此外,metaSPAdes 创建一致序列以形成每个重叠群的高质量代表,而不是试图代表每个菌株。MEGAHIT 被发现适用于以基因组和基因为中心的研究,如果可用的计算资源较少,则应使用。一项将不同组装软件应用于 Tara Ocean 宏基因组数据集的研究发现,SPAdes 生成了最长的重叠群,这使其最适合以基因组为中心的研究 。SPAdes 和 IDBA-UD 还导致了与参考基因组不对齐的最大数量的错误组装和重叠群。关于计算资源,同一项研究发现 SPAdes 是“内存最昂贵”的,而 MEGAHIT 是内存效率最高的。此外,IDBA-UD、SPAdes 和 metaSPAdes 需要最长的运行时间。另一项研究评估了个组装器在两个模拟宏基因组数据集上的性能:一个具有低丰度物种的高多样性数据集,类似于土壤等环境,以及一个具有密切相关物种的低多样性数据集,它模拟了生态系统,例如生物膜,以及低复杂度的真实宏基因组。他们根据与组装数量大小、组装精度和组装完整性相关的指标对最终组装进行了评估。对于模拟和真实的低复杂度宏基因组,发现 MaSuRCA 是最好的组装器,其次是 metaSPAdes。对于高度复杂的宏基因组,最好的组装器是 metaSPAdes。MEGAHIT 被发现适用于保守的组装策略,并且被发现具有低错误率和低 N50。鉴于肠道和皮肤是不同的样本,并且有足够的计算资源可用,因此本方法中使用了 metaSPAdes 进行组装。
第 3 阶段:分箱(第 17 步)
宏基因组组装高度碎片化,很难先验地确定重叠群源自哪个基因组,甚至样本中存在多少基因组。“分箱”的目标是对组装软件输出的支架或重叠群进行聚类,以生成分箱或假定的基因组草图。存在许多不同的工具来执行基因组分箱。这些工具可以根据它们提取用于执行分箱的特征的策略进行划分,包括基于序列组成的方法、基于序列丰度的方法和混合方法。基于序列组成的方法依赖于诸如 k-mer 核苷酸频率(样本中检测到的长度为 k 的所有序列组合的频率)和 G/C 含量等指标。基于丰度的方法可以处理一个样本或多个样本。适用于多个样本的方法依赖于这样的假设:来自相同基因组的重叠群的覆盖范围应该在不同样本之间相关。混合分箱方法,例如 CONCOCT、MetaBat 2和 MaxBin 2,同时考虑了序列组成和丰度,并且已被证明可以实现更准确的分箱。分箱算法可以显示不同样本甚至一个样本中不同群落的不同性能。DAS Tool 和 MetaWRAP 等 bin 合并工具可用于平衡不同分箱方法的优势和劣势,并通过组合许多不同分箱工具的输出并选择最佳分箱来恢复最佳质量 的分箱。在此方法中,CONCOCT、MetaBat 2 和 MaxBin 2通过 MetaWRAP 流程使用。
第 4 阶段:质量评估和分箱提纯(步骤 18-26)
从宏基因组数据中恢复 MAG 是一个复杂的问题,需要谨慎和仔细的校正。因此,在对分类、系统发育和/或功能潜力进行推断之前,评估分箱的质量非常重要。分箱的质量评估包括诸如完整性(包含在 bin 中的基因组的估计比例)、污染率(来自其他生物体的基因组序列的数量,被错误地与基因组草图合并)和菌株异质性等指标。通常通过检测基因组草图中单拷贝标记基因的存在和数量来估计完整性和污染率。未受污染且完整的 MAG 将在基因组中仅存在一次所有这些标记基因。在该方法中,使用了最广泛使用的用于估计原核基因组完整性/污染率的工具之一 CheckM(图 3a、b)。CheckM 通过使用一组谱系特异性标记基因来评估基因组的质量,这些标记基因是通过将每个基因组放置在预先计算的系统发育参考树中来确定的。可以在本方法中使用的 CMSeq 流程 中实施的宏基因组读长比对来估计株水平异质性(图 3c)。CMSeq 通过基于比对结果计算 MAG 中检测到的多态率(非同义替换的比例)来估计株异质性。使用这些指标,可以将 MAG 分配到不同的质量级别。根据关于宏基因组组装基因组 (MIMAG) 的最低信息标准,完整性 >90% 和 <5% 污染的 MAG 应报告为高质量的基因组草图。这些高质量的基因组还应包含 5S、16S 和 23S rRNA 基因,以及 20 种可编码氨基酸中至少 18 种的 tRNA。然而,如之前所示rRNA 操纵子是从鸟枪法宏基因组测序数据组装的一个具有挑战性的区域。完整性≥50% 且污染<10% 的MAG 应报告为中等质量。所有其他具有 <50% 完整性和 <10% 污染的 MAG 都应标记为低质量基因组。由于污染估计对于更不完整的基因组不太可靠,因此可以估计一个额外的质量分数 (QS),计算为完整性 –(5 × 污染)以进行进一步的 MAG 质量过滤。关于株异质性,0.5% 的阈值先前已被证明是区分高质量和中等质量 MAG 的可靠指标。另一种评估 MAG 质量的方法是通过全基因组比对和与参考分离基因组的比较(图 3d、e)。要将 MAG 与 RefSeq 或内部基因组集合进行比较,可以使用 Mash或 MUMmer 等软件工具,本方法中均展示了这些工具。
图 3:原核 MAG 的质量评估和物种分类
Fig. 3: Quality assessment and taxonomic classification of prokaryotic MAGs.
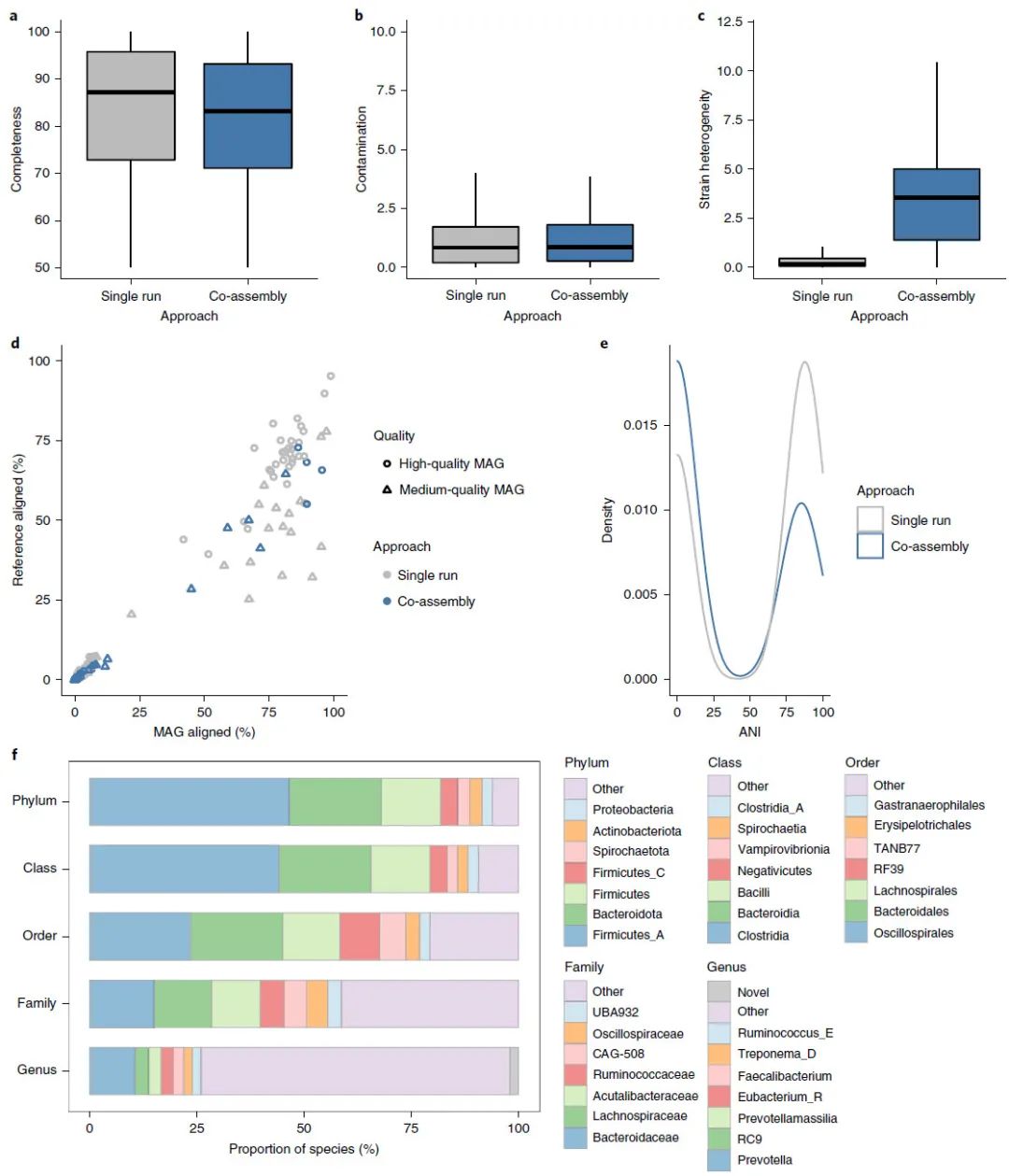
a,完整性,b,污染率,c,我们的单样本和共同组装的 MAG 的株异质性。与单样本的 MAG 相比,共同组装的 MAG 具有较低的完整性和较高的株异质性。d,比对的 MAG 百分比与针对不同组装方法和 MAG 质量比对的分离(参考)基因组百分比。鉴于预计 MAG 不如参考基因组完整,预计 MAG 比对的百分比将高于分离物比对的百分比。应使用 Anvi’o 等工具进一步检查违反此预期的 MAG。e,这些 MAG 菌株的 ANI 分布比较。以下对应于所考虑的每种方法的 MAG 数量:单样本:n = 422,共同组装:n = 175。框长度表示数据的 IQR,并且须延伸到 1.5 × IQR 内的最低和最高值分别来自第一四分位数和第三四分位数。与使用 GTDB-Tk 对我们的 249 个去冗余单样本和共组装的MAG 进行物种分类相比,单样本 MAG 似乎与参考基因组具有更高的比对同一性。分类单元名称代表来自 GTDB r95 的名称。通过我们的分析,可以恢复灰色阴影的新型(未培养的)生物。
然后可以使用这些质量估计来提高分箱的质量。更具体地说,在当前的工作流程中,我们使用 MetaWRAP分箱 提纯,它使用 CheckM 完整性和污染估计来组合来自多个原始 分箱预测的输出,并使用“bin_refinement”函数生成改进的 分箱集。这个提纯过程首先生成混合分箱集,确保如果两个重叠群在任何原始集中的不同分箱中,它们不会放在混合分箱中。接下来,“bin_refinement”功能会考虑原始分箱 集以及不同的混合变体,以使用 CheckM 完整性和污染率估计来选择最佳质量的分箱。
虽然本方法中没有描述,但可以使用各种工具通过手动校正和重叠群分类对 MAG 集进行质量控制。MAG 可以使用 Anvi’o 等方法手动管理,该方法允许用户目视检查序列组成和差异覆盖的模式,以手动删除这些指标不同的重叠群。更大规模的研究可以受益于 RefineM 等工具,它可以自动识别具有不同四核苷酸特征、覆盖率或 GC 含量的重叠群。它还对基因组内的重叠群进行分类以去除潜在的异常值。Contig Annotation Tool (CAT) 可以类似地用于对每个重叠群进行分类并去除系统发育不一致的序列。然而,这些方法的应用受到不完整的参考数据库和潜在的不正确标记和/或参考基因组污染的限制。
第 5 阶段:MAG 的去冗余(步骤 27)
在对分箱进行质量控制和提纯之后,可选的下一步是基因组去分箱。去分箱是对具有高序列相似性的基因组进行聚类,并为每个聚类确定最高质量的代表性基因组的过程。是否去冗余的选择取决于科学问题和研究人员可用的计算资源。去冗余有很多优点。基因组去冗余减少了下游分析的计算负担。它还提高了下游步骤(例如读长比对)的整体准确性。将测序读数比对回基因组集时,基因组集中的高度冗余会导致读长随机分配给多个冗余基因组,从而导致对冗余基因组的人为低丰度估计。然而,如果分析的目标是更好地了解种群多样性、进化或阐明附属基因组,则考虑整个基因组集很重要。在去冗余的过程中,需要进行成对的基因组比较,并且基因组比较的数量与基因组数量成二次方增加。可以使用各种度量来估计两个基因组之间的相似性,包括通过基因组的核苷酸水平比较估计的平均核苷酸同一性 (ANI) 和通过比较相应氨基酸估计的平均氨基酸同一性 (AAI)。ANI 用于估计密切相关基因组(物种或菌株水平比较)之间的基因组相似性,而 AAI 可用于估计更远相关基因组之间的相似性。本方法中使用的 dRep 等工具首先使用 Mash 对基因组距离进行快速估计,以形成大致相似基因组的主要集群,然后通过计算主要集群的每个成员之间的 ANI 来更准确地测量基因组距离。Mash的准确性对基因组大小和低完整性水平之间的巨大差异很敏感,因此在比较具有相似长度且完整性至少为 50% 的基因组时,该方法最为准确。次级簇的阈值取决于要解决的问题。90,000 个原核基因组的成对 ANI 比较发现细菌物种边界通常位于 ~95% 的 ANI。因此,如果目标是在物种水平上了解 MAG 的多样性,则可以使用 95% 的 ANI 阈值。在比较两个基因组时,比对分数 (AF) 也是一个需要考虑的重要因素。例如,通过水平基因转移,两个基因组可以共享一个小的可移动遗传元件,导致高 ANI 但低比对分数。有人提议 60% 的 AF 可用于物种级别的划分。对于不完整的 MAG,该值可以说是过于严格,因为 MAG 的较低 AF 足以分配物种级别的匹配(例如,Almeida 等人使用了30% 的 AF)。在形成冗余基因组的二级簇后,dRep 使用各种指标(如 N50、基因组长度以及 CheckM 对完整性、污染和菌株异质性的估计)从每个二级簇中选择最佳基因组。
阶段 6:物种分类(步骤 28)
可以使用多种方法对基因组进行准确的分类,其中大部分方法依赖于与参考数据库(如 NCBI GenBank 或基因组分类数据库 (GTDB))的序列比较。例如,基因组分类数据库工具包 (GTDB-Tk)利用细菌和古菌参考树和多序列比对,以及 GTDB 分类框架为每个基因组分配分类谱系。为了对基因组进行分类,GTDB 流程首先使用 Prodigal 预测蛋白质编码序列,并使用 HMMER 在基因组草图中检测一组 >100 个预定义的细菌和古菌标记基因。然后通过 pplacer 的系统发育放置将基因组分配参考树中的特定区域。分配给基因组的分类法取决于其在 GTDB 参考树中的位置、其相对进化分歧 (RED) 分数及其与最接近的参考基因组相关的 ANI。在该方法中,GTDB 和 GTDB-Tk 用于 MAG 的物种分类(图 3f)。
阶段 7:瓶颈评估(步骤 29-34)
我们可以通过将读长比对回组装和基因组集来估计捕获的基因组多样性的数量。对于每次运行,我们在 y 轴上绘制映射到其各自组装的读数百分比与映射到 x 轴上设置的最终基因组(MAG)的百分比读数(图 4a)。通过以这种方式可视化数据,可以判断映射到 MAG 的低百分比读数是否是由于与组装或分箱相关的问题。结果图中的底部象限代表未组装大部分序列读长的样本。各种因素会影响比对到组装集的读长百分比。一项研究使用 CAMISIM 微生物群落和宏基因组模拟器来创建不同的微生物群落,发现组装性能随着株水平多样性、异常错误分布和不均匀覆盖分布而降低。左上象限代表其中大部分读长已组装但重叠群未分箱或分箱对中等或高质量 MAG 没有贡献的样本。这些样本可能包含大量真核、病毒或质粒序列,这些序列可以组装但可能无法分箱以产生 MAG。右下角代表即使每个样本都没有成功组装的样本,我们可以在对使用其他样本生成的基因组进行比对时招募它们的大部分多样性。最后,右上角代表很好地映射到它们各自的组装集和最终 MAG 集的样本。该象限中的样本通常不太多样化。目标是获得映射到基因组集的高比例读长,例如使用不同的组装和分箱策略,这样无论比对到组装的读长百分比如何,我们都有高百分比的读长比对到最终 MAG 集。增加比对射到基因组集的读长百分比的一种方法是通过共同组装,如本方法所示。在共组装中,相关的宏基因组根据相关的元数据进行组合并组装在一起。此后,所有单独的宏基因组(这里称为运行)都被比对到共组装以执行分箱过程。通过这种方式,可以使用单个运行内的序列丰度和运行之间的差异丰度将重叠群聚类到分箱中。共组装特别有利于回收样品中低丰度存在的物种。
图 4:使用读长比对来可视化组装和分箱中的瓶颈。
Fig. 4: Using read mapping to visualize bottlenecks in assembly and binning.
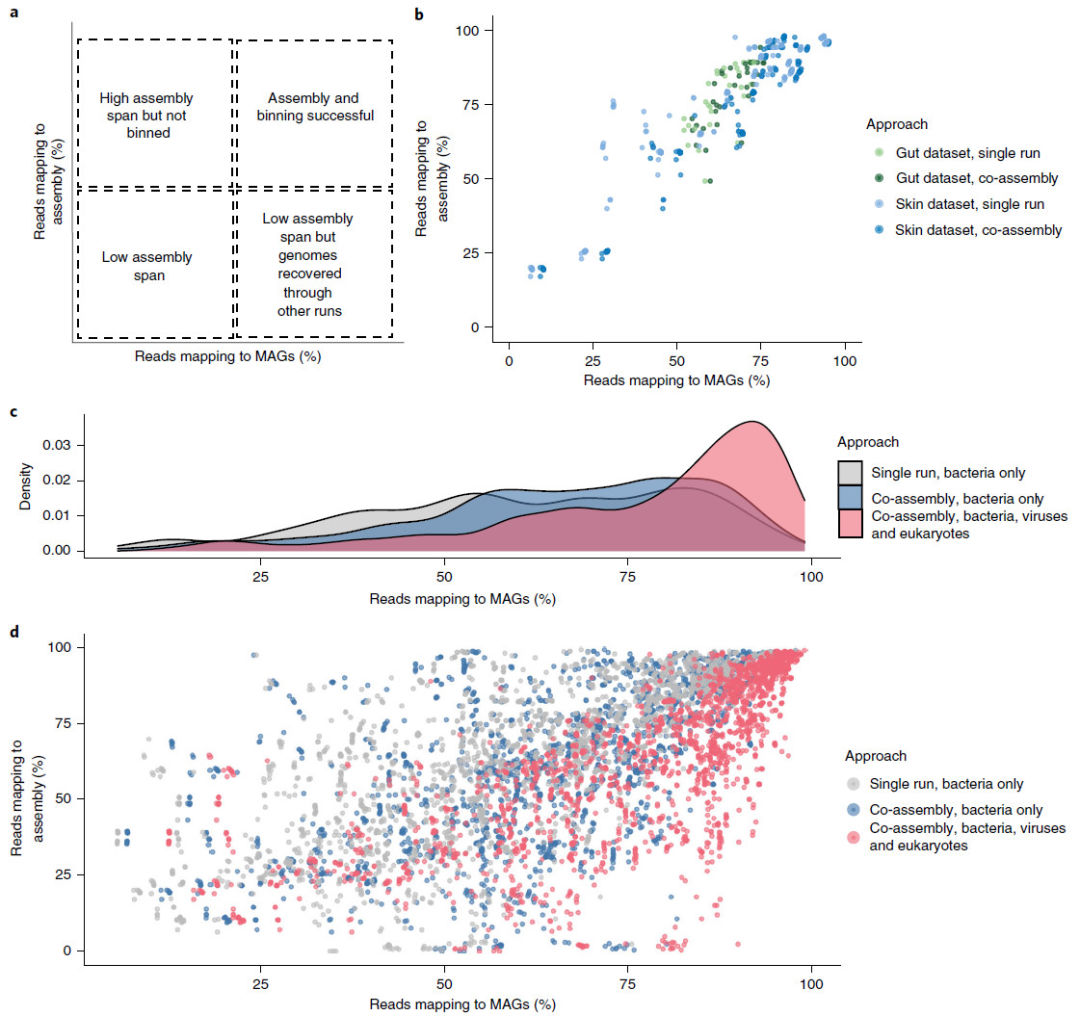
a,用于评估从鸟枪法宏基因组测序数据中恢复基因组的组装和分箱方法的框架。对于每次运行,我们绘制比对到我们去冗余的 MAG 的百分比读长与比对到该样本组装集的读长百分比。基于样本相对于四个象限的位置,可以确定从样本中恢复多样性的瓶颈。在左上象限中运行,其中有高百分比的读长比对到组装,但读长比对到 MAG 集的百分比低,是那些可以从不同的 分箱或真核和病毒表征中受益的人。在左下象限中运行,其中组装范围低且该样本中的多样性未捕获到 MAG 集中,例如,分析其他数据集,以便未通过一次样本捕获的生物可以通过另一个样本进行组装和分箱。右下象限中的点表示组装范围较低的样本,但通过其他样本捕获了多样性。右上象限中的运行是那些组装范围很大并且大部分多样性也被捕获到去冗余的 MAG 集中的样本。b,我们用本方法中调查的肠道数据集展示了这个框架,作为一个额外的例子,153 个人皮肤运行来自我们对健康人类皮肤微生物组(本方法未涵盖)的调查,它揭示了在本方法中发现的两个象限皮肤数据集但不在我们的肠道数据集中这与具有大量真核和病毒多样性的皮肤一致。c,在我们的皮肤研究中,共组装、真核和病毒分析对比对到基因组集的百分比读长影响的可视化。d,使用共组装,通过汇集共享相关元数据的样本,以及通过我们的真核和病毒分析,可以在我们的基因组集中加入额外的多样性。因此,即使在读长比对到组装集的百分比很低的情况下,我们也能够实现到 MAG 的高百分比映射。虽然该方法没有详细说明我们的皮肤分析,但数据集可以从 SRA 下载,项目编号 PRJNA46333。
流程和语言
在处理大型数据集时,工作流语言对于确保可重复性和可扩展性至关重要。我们的工作是作为模块化 Snakemake 流程 (https://github.com/Finn-Lab/MAG_Snakemake_wf) 提供的,并执行从 SRA 下载数据到生成图片的每一步,所有步骤都无需人工干预。步骤以及相关的 Snakemake 规则如图 1a 所示。运行我们的流程所期望的输出目录树如图 1b 所示。
材料
硬件
至少500GB内存的高性能计算机
软件
MAG Snakemake 流程 (https://github.com/Finn-Lab/MAG_Snakemake_wf)
Singularity 3.5.0 (https://github.com/hpcng/singularity)
Snakemake 版本 5.18 (https://github.com/snakemake/snakemake)
运行 MAG Snakemake 流程将自动从 SRA 下载测序数据。
测序数据可以通过使用 SRA 运行加入手动下载:
SRR1761666
SRR1761673
SRR1761674
SRR1761675
SRR1761698
SRR1761700
SRR1761701
SRR1761702
SRR1761705
SRR1761706
SRR1761708
SRR1761710
SRR1761713
SRR1761714
SRR1761717
SRR1761718
SRR1761719
SRR1761720
SRR1927149
SRR1929484
SRR1929485
SRR1929563
SRR1929574
SRR1930122
SRR1930128
SRR1930132
SRR1930133
SRR1930134
SRR1930138
SRR1930140
SRR1930142
SRR1930143
SRR1930144
SRR1930149
SRR1930176
SRR1930177
SRR1930179
SRR1930187
SRR1930244
Snakemake流程还将下载相关的singularity容器,以便可以使用我们流程所需的相关软件。或者,可以从以下位置手动下载这些工具:
ncbi-genome-download version 0.3.0 (https://github.com/kblin/ncbi-genome-download)
Mash version 2.2.1 (https://github.com/marbl/Mash)
parallel-fastq-dump version 0.6.6 & fastq-dump version 2.8.0 (https://github.com/rvalieris/parallel-fastq-dump)
FastQC = 0.11.7 (https://github.com/s-andrews/FastQC)
MultiQC = 1.3 (https://github.com/ewels/MultiQC)
kneaddata = 0.7.4 with Trimmomatic = 0.39 & Bowtie = 2.4.2
metaSPAdes version 3.14.0 (https://github.com/ablab/spades)
MetaWRAP = version 1.2.2 (https://github.com/bxlab/metaWRAP)
CheckM version 1.0.12 (https://github.com/Ecogenomics/CheckM)
Bowtie version 2.4.1 (https://github.com/BenLangmead/bowtie2)
Prokka version 1.14.5 (https://github.com/tseemann/prokka)
CMSeq version 1.0 (https://bitbucket.org/CibioCM/cmseq)
MUMmer version 3.23 (https://github.com/mummer4/mummer)
dRep version 2.3.2 (https://github.com/MrOlm/drep)
GTDB-Tk version 1.3.0 (https://github.com/Ecogenomics/GTDBTk)
BWA version 0.7.17 (https://github.com/lh3/bwa)
samtools version 1.9 (https://github.com/samtools/samtools)
数据库
RefSeq完整细菌基因组 (downloaded May 2020; https://www.ncbi.nlm.nih.gov/refseq/)
GTDB数据库,版本95 (https://data.ace.uq.edu.au/public/gtdb/data/releases/)
CheckM数据库 (https://data.ace.uq.edu.au/public/CheckM_databases/)
步骤
流程安装
1 下载GTDB数据库95版(33G)
wget -c https://data.ace.uq.edu.au/public/gtdb/data/releases/release95/95.0/auxillary_files/gtdbtk_r95_data.tar.gz
关键步骤:GTDB 目前每两年更新一次,以包括 NCBI 组装数据库中存在的最新基因组
译者注:GTDB更新很频繁,2019年更新了4次(80/83/86/89),20年7月更新了95版,21年4月更新了202版(47G)。最新链接如下:
wget -c https://data.ace.uq.edu.au/public/gtdb/data/releases/release202/202.0/auxillary_files/gtdbtk_r202_data.tar.gz
2 下载RefSeq细菌基因组
ncbi-genome-download bacteria –formats fasta –p refseq –assembly-levels complete
其中“bacteria”和参数“-p refseq”确保从 NCBI RefSeq 存储库下载细菌基因组。此外,“-formats fasta”表示基因组应为fasta 格式,“-assembly-level complete”仅下载完整的细菌 RefSeq 基因组。
关键步骤:如果需要古细菌基因组,同样可以使用“archaea”选项从 RefSeq 下载它们。
关键步骤:该方法于 2020 年 5 月下载的 RefSeq。
3 接下来,使用默认 k-mer和sketch大小生成数据库的 Mash sketch:
mash sketch -o refseq.msh /path/to/RefSeq/*fasta
4 从 https://github.com/Finn-Lab/MAG_Snakemake_wf 将流程的代码下载到至少有 1.5 TB 磁盘空间的位置。将目录更改为该文件夹。使用以下命令将 GTDB 数据库和 RefSeq Mash 草图移动到子文件夹 /data/databases:
cd /path/to/MAG_Snakemake_wf/
mkdir -p data/databases
mv /path/to/refseq.msh data/databases
mv /path/to/gtdbtk_r95_data.tar.gz data/databases
tar -xvzf data/databases/gtdbtk_r95_data.tar.gz
**5 安装 Python 3 版本的 Miniconda (**http://conda.pydata.org/miniconda.html)。对是否应将 conda 放入 PATH 的问题回答“是”。获取 Singularity 版本 3.5.0。
wget -c https://repo.anaconda.com/miniconda/Miniconda3-py39_4.9.2-Linux-x86_64.sh
bash Miniconda3-py39_4.9.2-Linux-x86_64.sh
6 安装snakemake
conda create -c conda-forge -c bioconda -n snakemake snakemake=5.18
conda activate snakemake
7 使用runs.txt 文件(我们之前对人类肠道微生物组进行的34 个测序样本)指定要分析的SRA 运行样本,每行都有不同的样本加入。使用 coassembly_runs.txt 文件(两个联合组装样本由 14 次测序样本组成)指定将合并到每个共组装样本中的样本。
8 运行流程
LSF作业系统提交任务:
snakemake –use-singularity –restart-times 3 -k –jobs 50 –cluster-config clusterconfig.yaml –cluster “bsub -n {cluster.nCPU} -M {cluster.mem} -e {cluster.error} -o {cluster.output}”
SLURM作业系统提交任务:
snakemake –use-singularity –restart-times 3 -k -j 50 –cluster-config clusterconfig.yaml –cluster “sbatch -n {cluster.nCPU} –mem {cluster.mem} -e {cluster.error} -o {cluster.output}”
关键步骤:运行此流程应自动生成与此协议相关联的数字,而无需人工干预。方法的其余部分将介绍构成流程的不同步骤。
数据下载
计算时间:16小时
9 要使用 SRA 工具包下载与 SRA样本相关的测序数据,请使用:
prefetch {params.run} && vdb-validate {params.run} && parallel-fastq-dump –threads {threads} –outdir {params.outdir} –skip-technical –split-3 –sra-id {params.run} –gzip
Prefetch将保存与相关的 SRA 文件列表加入 {params.run} 。可以使用 vdb-config -i 设置存储 SRA 数据的路径,并将“location of user-repository”设置为具有足够存储空间的路径,然后通过键入“x”关闭交互窗口。检查 SRA 数据集的完整性以确保数据已成功下载至关重要。这可以使用 vdb-validate 来执行。接下来,使用 parallel-fastq-dump 将 SRA 文件转换为 FASTQ 格式。—split-3 选项将读长分成左右两端,但如果有不匹配的两端,这些将放在第三个单个文件。由于不需要诸如条形码和引物读长之类的技术读长,因此包含 —skip-technical 选项以删除这些技术部分。接下来,使用 —gzip 选项压缩生成的 FASTQ 文件。
预处理
kneaddata计算时间:48小时
10 评估原始数据的质量以确保它没有任何问题或偏差很重要。FastQC 生成一个报告,以可视化与测序仪或起始库相关的问题。FastQC 的一个重要警告是,某些模块(例如重复序列和过丰富序列)仅计算数据子集(即每个文件中的前 100,000 个不同序列)的统计数据。可以提供将针对文库进行搜索的附加接头序列。如果未设置此选项,将搜索一组有限的接头。要对样本的正向读长运行 FastQC,请使用:
fastqc {input.fwd} –outdir {params.outdir}
其中 {input.fwd} 是 FASTQ 格式(或 FASTQ gzipped 格式)的正向读长,{params.outdir} 是存储 FastQC 分析输出的文件夹。可以使用相同的程序在反向读长上运行 FastQC。
11 MultiQC 汇总多个样品的 FastQC 结果并创建单一报告以便于比较。MultiQC 报告提供有关整个数据集的信息,而 FastQC 报告可用于可视化单个读长的质量。要创建 MultiQC 报告,请使用:
multiqc {params.indir} -o {params.outdir}
在此命令中,{params.outdir} 表示输出目录,而 {params.indir} 是包含要聚合和汇总的所有 FastQC 分析的先前输出目录的目录。
12 接下来,创建一个净化数据库,KneadData 将在下游预处理分析中使用该数据库。KneadData 是一种分析 流程,可用于质量控制和去除宿主污染。要使用 KneadData,需要一个 Bowtie2 格式的索引污染物数据库和 FASTQ 格式(或 FASTQ gzipped 格式)的读长。要去除人类宿主污染,请提供人类基因组的参考以比对读长。要以 Bowtie2 格式下载预先编入索引的人类基因组,请使用以下命令:
kneaddata_database –download human_genome bowtie2 {params.outdir}
创建去污数据库时,重要的是要考虑 PhiX (https://www.ncbi.nlm.nih.gov/nuccore/9626372) 是否作为掺入添加到测序样品中,样品是否从宿主或实验室是否使用可能污染样品的其他生物体的 DNA。接下来,应将 PhiX、宿主和其他潜在污染物的基因组添加到一个文件中,并使用 Bowtie2 进行索引以创建污染物数据库。请注意,特别是对于低微生物生物量样品,例如皮肤拭子,还必须考虑试剂衍生的污染,通常称为“kitome(试剂组)”,因为即使是低水平的污染也会与已经微弱的“真实”信号竞争这些样品。解决试剂衍生污染的一种方法是通过合并和分析“空白”测序对照,其中试剂在不添加样品材料的情况下进行测序。通过阴性对照检测到的污染序列可以添加到污染物数据库中。
13 进行质控和去污染
kneaddata –remove-intermediate-output -t {threads} –input {input.fwd} –input {input.rev} –output {params.outdir} –reference-db {input.index} –trimmomatic {input.trimmomatic_path} –trimmomatic-options “ILLUMINACLIP:{input.pathtoadapters}:2:30:10: SLIDINGWINDOW:4:20 MINLEN:50” –bowtie2-options “–very-sensitive –dovetail”
其中 {input.fwd} 和 {input.rev} 是输入 FASTQ 文件,它们为双端数据提供了两次。{params.outdir} 是输出目录,{input.index} 是去污数据库文件夹的路径,{input.trimmomatic_path} 是包含 jarfile 的 Trimmomatic 文件夹的路径,{threads} 是分配的线程数.对于 —trimmomatic-options,使用‘ILLUMINACLIP:{input.pathtoadapters}:2:30:10: SLIDINGWINDOW:4:20 MINLEN:50’。ILLUMINACLIP 用于修剪接头和其他特定于Illumina的序列。使用“ILLUMINACLIP:{input.pathtoadapters}:2:30:10”,Trimmomatic 在文件 {input.pathtoadapters} 和每次读长中详述的接头之间搜索种子匹配或短对齐,允许最多两个不匹配。种子将被扩展和剪切,双端读长的分数为 30,单端读长的分数为 10。通常,读取质量在读长结束时会变差。使用选项 ‘SLIDINGWINDOW:4:20 MINLEN:50’ 修剪低质量的末端,当质量(也称为 Phred 分数)在此窗口中低于 20 时,将修剪 4 nt 的滑动窗口。接下来,删除任何序列短于 50 nt。在这里,参数的顺序很重要,因为在 SLIDINGWINDOW 之前添加 MINLEN25 只会在修剪前检查读取的长度,而在 SLIDINGWINDOW 之后添加 MINLEN25 会删除修剪后太短的读取。要使用的 —bowtie2-options 是“—very-sensitive”和“—dovetail”,用于将比对参数设置为非常敏感,并设置配对相互延伸以保持一致的情况。要减少分析所需的存储空间,请添加选项 —remove-intermediate-output 以便删除中间文件,包括大型 FASTQ。
修剪和过滤后的最终文件将分别作为 {inputname}_1_kneaddata_paired_1.fastq 和 {inputname}_1_kneaddata_paired_2.fastq 存储在输出目录中,用于正向和反向读长。这些文件将用于下游分析。kneaddata 的替代方案是基于 k-mer 的 BBDuk,它是 BBTools 包 (http://sourceforge.net/projects/bbmap/) 的一部分,可用于质量、接头剪切和污染物过滤。
关键步骤:对测序数据进行彻底的质量控制很重要,因为这一步会影响所有后续的下游分析。
14 接下来,重新运行 FastQC 和 MultiQC,如步骤 10 和 11 中所示,以可视化预处理对数据集的影响。图 2 显示了预处理后读长的平均质量分数和接头含量的 MultiQC 报告。
组装和共组装
CPU计算时间:2600i小时
15 要创建共组装示例,请按照文件 coassembly_runs.txt 中的详细说明将步骤 13 中预处理的 FASTQ 文件按元数据的分组合并,如下所示:
cat {input.fwd1_to_fwdN} > {output.coas_fwd}
其中 {input.fwd1_to_fwdN} 是属于由空格分隔的感兴趣元数据的运行的所有前向读长,{output.coas_fwd} 是这些运行的前向读长取。重复该过程以生成联合组装反向读长取。尽管本方法在聚合运行以创建联合组装样本时考虑了年龄元数据,但其他样本元数据字段(例如,在欧洲核苷酸档案中可用)也可用于对样本进行分组。
16 组装读长。可使用metaSPAdes(A)或MEGAHIT(B)
(A) MetaSPAdes
要使用 metaSPAdes,请按如下方式提供单样本和共组装双端读长:
spades.py –meta -1 {input.fwd} -2 {input.rev} -o {params.outdir} –threads {threads} -m {resources.mem}
其中 {threads} 是分配的线程数,-m 指定 SPAdes 的 RAM 限制(以 Gb 为单位),如果超过此限制,SPAdes 将终止。
(B) MEGAHIT
要使用 MEGAHIT,类似地指定共组装:
megahit -1 {input_1.fwd} -2 {input_1.rev} -o {params.outdir} -t {threads}
其中 -o 指定输出目录,{threads} 指定分配的线程数。MEGAHIT 生成的 final.contigs.fa 文件在标题(fasta 文件中以“>”开头的行)中有空格和特殊字符。使用以下方法可视化文件头:
head final.contigs.fa
这些序列ID必须在下游分析之前进行格式化。
分箱
CPU时间:100小时
17 宏基因组组装可以包含许多短重叠群,这些短重叠群本身在丰度和序列组成方面具有很大的变化。因此,这些短重叠群更有可能被错误地分箱。以前的工作发现,重叠群大小的合适下限是 2.5 kb,但如果组装质量非常好,则可以使用重叠群长度低至 1.5 kb。执行分箱如下:
metawrap binning -t {threads} -m {resources.mem} -a {input.fasta} –maxbin2 –metabat2 –concoct -l {params.mincontiglength} -o {params.outdir} {input.fwd} {input.rev}
其中,{input.fasta} 是通过组装获得的支架文件的路径,{params.mincontiglength} 表示最小重叠群长度并指定为 2500。输出文件夹将具有三个最终 bin 集:concoct_bins、maxbin2_bins、metabat2_bins。对于 co-binning,{input.fwd} 和 {input.rev} 表示正向和反向读长,每个读长取都由一个空格分隔。
质量评估和分箱提纯:估计 MAG 的完整性和污染率
CPU时间:25小时
18 CheckM 可用于评估分箱的完整性和污染程度。CheckM 通过几个步骤评估分箱的质量。首先,“tree”命令将基因组分箱放入参考基因组树中。“lineage_set”命令创建一个标记文件,显示可用于评估每个 分箱的特定于谱系的标记集。然后将此标记文件传递给“analyze”,它识别将用于估计每个分箱的完整性和污染率的标记基因。‘qa’命令总结了不同汇总表中每个分箱的质量。使用以下命令执行上述步骤:
checkm lineage_wf -t {threads} -x {params.ext} –tab_table -f {output} {params.indir} {params.outdir}
其中 {threads} 表示线程数,{params.indir} 是包含所有分箱的目录,{params.ext} 表示分箱’fa’ 的扩展名,{params.outdir} 是 CheckM 输出目录和 {output} 是摘要 tsv 文件。
关键步骤:分箱的质量控制至关重要,因为包含低质量(高度碎片化、不完整和/或受污染)的基因组将导致误导性的物种和功能表征。
质量评估和分箱提纯:估计 MAG 的株异质性
CPU时间:700小时
19 要计算株异质性,首先按如下方式索引 MAG FASTA 文件:
bowtie2-build {input.MAG} {params.prefix}
其中 {input.MAG} 是 MAG 的 FASTA 文件,{params.prefix} 是为生成的索引文件选择的命名前缀。
20 接下来,比对读长至MAG索引结果如下:
bowtie2 –very-sensitive-local -x {params.outprefix} -p {threads} -1 {input.fwd} -2 {input.rev} | samtools view -uS - | samtools sort {threads} - -o {params.outprefix}.bam
samtools index -@ {threads} {params.outprefix}.bam
其中 {input.fwd} 和 {input.rev} 是正向和反向读长,{threads} 是分配的线程数,{params.outprefix} 是用于命名输出文件的前缀。
21 细菌MAG运行Prokka:
prokka {input} –kingdom Bacteria –outdir {params.outdir} –prefix {params.prefix} –force –locustag {params.prefix} –cpus {threads}
其中 {input} 是 MAG FASTA 文件,{params.outdir} 是输出目录的名称,{params.prefix} 是标记输出文件时使用的名称。
关键步骤:使用选项 —kingdom Archaea 可以对古细菌 MAG 使用类似的程序。
22 接下来,检查多态位点并使用CMSeq解析非同义位置、同义位置和比对位置的数量,如下所示:
scripts/polymut.py -c {input.MAG} {params.outprefix}.bam –gff_file {input.gff} –mincov 10 –minqual 30 –dominant_frq_thrsh 0.8 >
{params.outprefix}.cmseq.csv
其中 {input.MAG} 是 MAG fasta 文件,{input.gff} 是 Prokka 在上一步中输出的扩展名为 gff 的文件,{params.outprefix} 是输出文件的前缀。
质量评估和分箱提纯;MAG 与 RefSeq 基因组的比较
CPU时间:1小时
23 另一种评估 MAG 质量的方法是将它们与分离基因组(例如 RefSeq 中的基因组)进行比较。为此,首先使用以下方法确定 MAG 与每个参考基因组之间的混合距离:
mash dist -p {threads} {input.db} {input.MAG} > {output}
其中 {threads} 是线程数,{input.db} 是 mash 数据库的路径,{input.MAG} 是将与 RefSeq 数据库进行比较的 MAG。
24 接下来,对 Mash 结果进行排序以确定最高命中及其 P 值,如下所示:
sort -gk3 {input} | sed -n 1p > {output}
其中 {input} 是来自上一步的 mash 距离文件。{output} 文件是一个制表符分隔的文件,其中的列代表参考基因组的路径、MAG 基因组的路径、混搭距离、相关的 P 值和匹配的哈希值。
关键步骤:考虑 P 值很重要,因为它们表示偶然看到给定混搭距离的概率。较低的 P 值表示对获得的混合距离估计有更大的信心。
25 接下来,使用以下方法计算 MAG 和此最佳混合命中之间的对齐百分比 ANI:
dnadiff {params.reference} {params.MAG} -p {params.name}
其中 {params.reference} 和 {params.MAG} 是 MAG fasta 文件和参考基因组 fasta 文件的路径。带有前缀 {paramas.name} 的感兴趣的输出文件具有扩展名“.report”,并总结了比对、差异和 SNP。要使用的值是与参考 (REF) 对齐的碱基百分比、与 MAG (QRY) 和 ANI (AvgIdentity) 对齐的碱基百分比。
质量评估和分箱提纯;箱提纯
CPU时间:260小时
26 接下来,使用 metaWRAP 细化 bin,如下所示:
metawrap bin_refinement -o {params.outdir} -t {threads} -A {params.metabat} -B {params.maxbin} -C {params.concoct} -c 50 -x 10
其中 {threads} 是线程数,输出目录中的 {params.outdir} 和 {params.metabat}、{params.maxbin} 和 {params.concoct} 是通过获得的 MetaBAT、MaxBin 和 CONCOCT 的路径分别分箱。-c 和 -x 可分别用于指定下游分析中所需的分箱质量。提纯后的分箱 将存储在 {params.outdir}/metawrap_50_10_bins 文件夹中。
关键步骤:此外,每个 pplacer 线程使用 > 40 GB 的内存,因此为了优化资源使用,分配的内存量(以 GB 为单位)应该比线程数大 40 倍。
MAG 的去冗余(可选)
CPU时间:4小时
27 各种阈值可用于去除基因组目录中的冗余。在这一步中,使用 dRep 以 95% 的 ANI 对基因组进行聚类,这是描述原核物种的常用阈值。首先,dRep 使用 Mash 将 MAG 分组为主要集群(约 90% ANI)。此后,通过比对基因组(默认情况下是通过 ANImf)将来自每个主要集群的 MAG 放入物种级别的次要集群 (95% ANI)。要去冗余 MAG,请使用以下命令:
dRep dereplicate -p {threads} {params.outdir} -g {params.indir}/*.fa -pa 0.90 -sa 0.95 -nc 0.30 -cm larger –genomeInfo {input.checkm} -comp 50 -con 5
其中 {params.indir} 是包含要删除的 MAG 的目录,{input.checkm} 是一个.csv 文件,包含四列,包括 bin 名称、完整性、污染和应变异质性以及标题“genome”、“completeness”、 “contamination”和“strain_heterogeneity”。此外,-nc 是二级比较期间基因组之间的最小重叠水平,-cm 是覆盖率方法,“larger”指定覆盖率应计算为两个基因组对齐部分的最大值:max((对齐长度/基因组 1),(aligned_length/基因组 2))。
关键步骤:鉴于 MAG 是不完整的,请使用 0.30 的覆盖阈值(参数 -nc),这不如施加在完整基因组上的 0.60 对齐分数那么严格。值得注意的是,这可能低估了物种代表的数量。
物种分类
CPU时间:3小时
28 要对细菌和古菌 MAG 进行分类,请使用基因组分类数据库 和基因组分类数据库工具包 (GTDB-Tk),如下所示。GTDB-Tk 指南建议该工具应用于具有 ≥ 50% 完整性和 ≤ 10% 污染的基因组。
gtdbtk classify_wf –cpus {threads} –genome_dir {params.indir} –out_dir {params.outdir} -x {params.ext}
其中 {params.indir} 是包含基因组的输入目录,{params.outdir} 是 GTDB 分析的输出目录,{params.ext} 是“fa”,因为分箱 具有“fa”扩展名。细菌的分类分析将存储在文件 gtdbtk.bac120.summary.tsv 中,古细菌的分类分析将存储在文件 gtdbtk.ar122.summary.tsv 中。
关键步骤:GTDB-Tk 需要多个 CPU 和 ≥ 128 GB 的 RAM。
瓶颈评估
CPU时间:120小时
29 将测序读长比对到参考基因组的过程需要参考的索引文件。要创建参考索引,请使用“bwa index”,如下所示:
bwa index {params.scaffold}
30 接下来,将预处理的测序读长比对到参考索引。BWA 的替代品是 Bowtie2。BWA 和 Bowtie2 的输出是 SAM 格式的比对文件。不是保存 bwa mem 的输出,而是通过“piping”将其重定向到 samtools 程序,立即将其转换为排序(二进制)BAM 格式。排序的过程涉及按照参考序列的顺序对比对进行排序。接下来,索引已排序的 BAM 文件。例如,要计算每个样本的读长比对到组装集的百分比,请使用
bwa mem -t {threads} {params.scaffold} {input.fwd} {input.rev} | samtools view -bS - | samtools sort -@ {threads} -o {params.alignedsorted} -
samtools index {params.alignedsorted}
samtools flagstat {params.alignedsorted} > {output.flagstat}
其中 {input.fwd} 和 {input.rev} 是正向和反向读取,-bS 表示输入是 SAM 格式,输出将是 BAM 格式,{params.alignedsorted} 是输出对齐方式 排序和索引。两个 samtools 命令的“-”表示输入是来自“标准输入”而不是来自文件。-o 是输出文件的名称。
31 要计算比对到基因组集的读取百分比,首先连接去冗余每个 MAG 的 fasta 文件:
cat {input.fasta1_to_fastaN} > {output.catalog}
其中 {input.fasta1_to_fastaN} 是 fasta 文件的路径,每个文件用空格分隔,{output.catalog} 是生成的 multifasta 文件。
32 然后按照与步骤 29 和 30 类似的过程进行操作:
bwa index {input.catalog}
bwa mem -t {threads} {input.catalog} {input.fwd}
{input.rev} | samtools view -bS - | samtools sort -@
{threads} -o {params.alignedsorted} -
samtools index {params.alignedsorted}
samtools flagstat {params.alignedsorted} > {output.flagstat}
其中 {input.catalog} 是步骤 31 中生成的 multifasta 文件({output.catalog})。
33 要使用步骤 30 和 32 中生成的 flagstat 输出 {output.flagstat} 计算映射到基因组目录或组集的读长百分比,请计算主要比对的数量(比对 - 次要 - 补充)。然后将主要对齐的数量除以正向和反向读取的数量,以获得映射的读长分数。
34 接下来,绘制比对到基因组数据库的读长百分比和比对到 y 轴上的组装集的读取百分比(参见图 4 中的示例)。
常见问题
步骤 问题 可能原因 解决方法
10,14 FastQC报告中‘Per sequence quality scores’ 显示WARNING或FAIL 质量分数均值<27显示WARNING,<20显示FAIL,大量的读长低质量、表明数据存在系统问题 通过质控剪切来缓解此问题
10,14 FastQC报告中‘Per base sequence content’ 显示WARNING或FAIL 给定一个随机库,预计碱基比例应该没有差异。如果 C&G 或 A&T 之间的差异在任何位置大于 10%,则发出警告,如果差异大于20%,则发出失败。巨大的差异可能表明存在未修剪的接头或有偏差的文库组成 如果根本原因是未修剪的接头,请将其移除
16 MetaSPAdes内存不足 MetaSPAdes 可能由于各种原因而失败,通常是由于内存分配不足。通过在 metaSPAdes 运行失败后运行命令 dmesg -T,我们可以更好地辨别失败的根本原因(例如,如果程序确实由于缺乏内存资源而被杀死) 可以将选项 —continue 或 —restart-from与相同的输出目录一起使用。—continue 选项使用相同的选项从最后一个可用的检查点运行 SPAdes,而 —restart-from 允许您更改某些组装选项
18 ls: 无法访问 storage/tree/concatenated.tre: 没有那个文件或目录 没有足够的 RAM 内存分配给 CheckM 增加对 CheckM 的内存分配
26 “运行 CheckM 时出了点问题。退出’ binning_refiner 失败可能是因为 bin 太少了 手动选择具有最佳 CheckM 完整性和污染估计的箱
预期结果
该方法的输出是包含一组基因组草图的 FASTA 文件,如果不需要去冗余,则由步骤 26 输出,或者在带有去冗余的情况下由步骤 27 输出,就像本方法的情况一样。在使用这些基因组草图进行下游分析之前,应该进行彻底的质量控制。例如,评估我们的 MAG 的一种方法是通过完整性、污染和菌株异质性的指标,如步骤 18-22(图 3)所示。值得注意的是,基于通用单拷贝标记基因的 CheckM 等工具依赖于参考数据库,因此可能无法对尚未纳入这些数据库的新型基因组的完整性提供可靠的估计。此外,并非每个重叠群都包含单拷贝标记基因,因此在某些情况下,污染可能无法检测到。克服这些限制并进一步验证 MAG 的一种方法是将它们与密切相关的分离基因组进行比较,如步骤 23-25(图 3d、e)所示,通过绘制比对的分离株与MAG 的 ANI百分比。鉴于预计 MAG 不如参考基因组完整,因此预计 MAG 比对的百分比通常高于比对的分离株的百分比。通过将 MAG 与分离株基因组对齐,可以确定 MAG 中缺少哪些基因组区域,但很难确定 MAG 或分离株未对齐的部分是由于 MAG 重建问题还是由于生物体的生物变异。完成质量评估后,可以将中等和高质量的单样本和共组装 MAG 一起去冗余以生成非冗余 MAG 集,如步骤 27 所示。使用 GTDB 对我们去冗余 的细菌 MAG 进行物种分类使用步骤 28 执行的操作总结在图 3f 中。
比对到我们的 MAG 的读长百分比可用于估计从样本中恢复的多样性比例。图 4b、d 中散点图上的每个点表示针对我们整个去冗余 MAG 集(x 轴)的样本比对读长的输出,以及针对该样本的相应宏基因组组装(y 轴)的比对读长。将我们的皮肤数据集与肠道数据集(图 4a)进行比较显示,皮肤有两个象限未被肠道数据集覆盖(左下角和左上角;图 4b)。在皮肤中发现的左下象限可能是由于皮肤中发现的高水平菌株异质性的结果,这导致组装的更大碎片并阻碍这些基因组的恢复,导致组装跨度低(低百分比读长比对到组装集)。左上象限中的样本可能代表那些已组装但质量不足以被招募到基因组集中的运行。左上象限中的运行还可以包括那些包含病毒、质粒或真核生物多样性的样品。这与具有相当大的真核生物种群的皮肤和许多偏向于原核生物基因组的分箱算法一致。为了解决这个问题,VIRify (https://github.com/EBI-Metagenomics/emg-viral-pipeline) 和 EukCC 等流程可分别用于恢复和表征病毒和真核序列。我们对皮肤研究的调查结果如图 4c、d 所示,用于单次组装,然后是共组装,然后是真核和病毒分析。共组装可用于捕获额外的多样性,但重要的是要注意,产生的基因组应被视为群体基因组,与单次运行的 MAG 相比,共组装的 MAG 存在更高的菌株异质性说明了这一点(图3c)。此外,重要的是要考虑根据元数据将哪些样本共同组装在一起,因为组装非常不同的样本可能会降低结果组装的质量。
该方法概述了从鸟枪宏基因组测序数据中回收和评估 MAG 的方法。可以使用以下指南 (https://ena-docs.readthedocs.io/en) 将产生的 QS > 50(质量分数 = 完整性 –(5 × 污染))、基因组箱和相关组件的 MAG 存放到 ENA(MAG提交指南) https://ena-docs.readthedocs.io/en/latest/submit/assembly/metagenome/mag.html# )。MAG 可用于各种下游应用。例如,一种应用是对基因组进行分类分析以发现新的生命进化枝(图 3f)。另一个应用是针对 MAG 进行读长比对,以使用Anvi’o 和 inStrain 等工具确定样本内或样本间 MAG 的丰度和基因组异质性。还可以对 MAG 进行功能分析,类似于 Almeida 等人的工作,以深入了解不可培养生物基因组中发现的基因和途径。最后,MAG 可用于补充参考数据库,以创建更全面的基因组集,当读长比对时,可以提供更大的分辨率,了解微生物在健康与疾病中的模式。
数据
图中显示的数据。2 和 3 可在支持的主要研究论文中找到。肠道的原始数据文件在图 3 和图 2-4 可作为项目加入 PRJNA268964 和 PRJNA278393 的一部分。图 4 中皮肤样本的原始数据文件可通过项目登录 PRJNA46333 获得。为该协议生成的数据集 2-4 可在 https://github.com/Finn-Lab/MAG_Snakemake_wf 子文件夹 Anticipated_Results 下找到。
代码
本方法中使用的代码可在 https://github.com/Finn-Lab/MAG_Snakemake_wf 公开获得。本方法中的代码已经过同行评审。
参考文献
Sara Saheb Kashaf, Alexandre Almeida, Julia A. Segre, Robert D. Finn. (2021). Recovering prokaryotic genomes from host-associated, short-read shotgun metagenomic sequencing data. Nature Protocols 16 2520-2541 doi: 10.1038/s41596-021-00508-2
- 本文作者: Anderson
- 本文链接: http://nikolahuang.github.io/2021/11/03/Nature子刊:宏基因组中挖掘原核基因组的分析流程/
- 版权声明: 转载请注明出处,谢谢。