

[TOC]
MyNotes:
总体概括为:全外显子测序的标准分析流程通常包括以下步骤:
质量控制(Quality Control,QC)
对原始测序数据进行质量控制,包括去除低质量的序列片段、过滤掉低质量的碱基和低覆盖度的测序片段等。
读取比对(Read Alignment)
将过滤后的测序数据比对到参考基因组上,以确定每个片段的位置和方向。
变异检测(Variant Calling)
通过对比测序数据和参考基因组,识别出样本中存在的单核苷酸多态性(Single Nucleotide Polymorphism,SNP)和插入/删除多态性(Insertion/Deletion,InDel)等变异。
注释(Annotation)
对检测到的变异进行注释,包括位置信息、功能影响、已知疾病关联等。
过滤(Filtering)
根据预设的过滤标准,过滤掉可能为假阳性的变异,如常见的多态性、测序误差等。
数据解读(Data Interpretation)
将过滤后的变异进行进一步分析和解读,以确定与研究对象相关的变异。
功能验证(Functional Validation)
通过实验验证已识别的变异是否与疾病相关,例如验证变异对基因表达、蛋白质功能的影响等。
分析流程
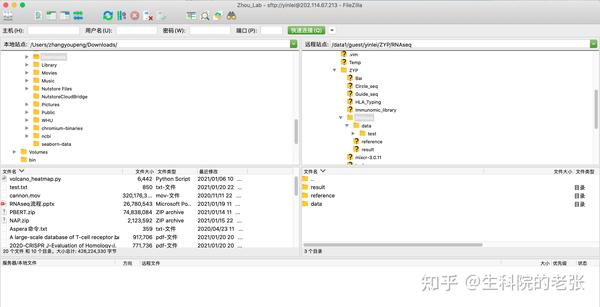
从本地硬盘通过FileZilla软件或在云端通过scp协议将所需数据传输至进行分析的服务器.
- FileZilla示例:
如下图所示,在窗口拖动数据文件到指定目录即可。
- scp协议示例:
命令行输入以下命令进行文件传输。
1 | scp -r local_folder remote_user_name@remote_ip:remote_folder |
- 先在服务器上安装conda, conda的安装使用请参照之前的文章生科院的老张:无root权限conda安装PyRosetta同源建模,安装好后通过conda进一步安装软件fastqc, multiqc, trimmomatic, bwa, samtools, bcftools, vcftools, gffread, 参考代码如下:
1 | conda create -n WES_WGS_WGRS #新建一个名为WES_WGS_WGRS的conda环境 |
- 如下图所示,ANNOVAR软件需要在官网 Download ANNOVAR - ANNOVAR Documentation 通过教育邮箱申请下载链接下载。

- 通过wget下载安装gtfToGenePred:
1 | wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64.v369/gtfToGenePred |
- 在R中安装stringr, circlize, grid, ComplexHeatmap, 需先安装Bioconductor, 详见之前的文章生科院的老张:RNAseq转录组差异表达分析教程:
1 | BiocManager::install('stringr') |
参考基因组和基因组注释文件:
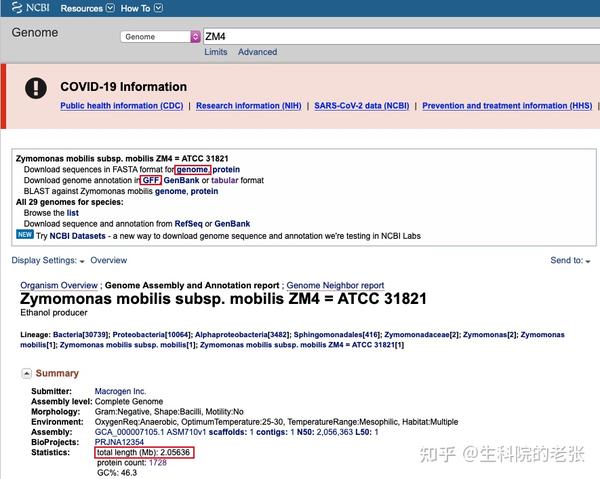
在对应的网站下载参考基因组和基因组注释文件, 如在NCBI的genome搜索ZM4能找到我要的物种对应的reference genome和annotation.
如图所示,点击红色方框内的genome和GFF会分别下载到.fasta格式的参考基因组序列和.gff格式的基因组注释。下方的基因组全长可以记录下来,在之后的分析中会使用到。
任何组学分析第一步都是对测序数据进行质量控制,细节可以参考之前的转录组教程 生科院的老张:RNAseq转录组差异表达分析教程 , 和转录组一样,使用md5检查数据完整性,fastqc进行质量检测,multiqc合并报告,trimmomatic进行质量过滤,参考代码如下:
1 | md5sum *gz > md5.txt && md5sum -c md5.txt |
2.2.2 fastqc质量控制与multiqc合并质控报告:
1 | fastqc *gz && multiqc ./ |
2.2.3 trimmomatic质量过滤:
1 | trimmomatic |
由于一批测序数据中有多个测序文件,因此需要循环以上过程,参考代码如下图所示:
1 | for dir in A B C D; #循环读取A,B,C,D四个目录 |
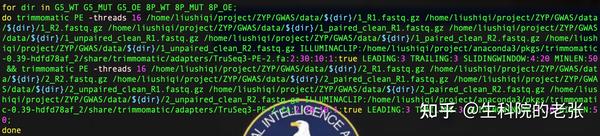
trimmomatic质量过滤
将经过质量控制的序列与参考基因组进行比对,参考代码如下:
1 | 建立比对索引 |
同样由于一批测序数据中有多个测序文件,因此需要循环以上过程,参考代码如下图所示:
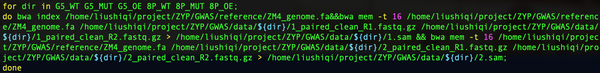
bwa比对基因组
1 | 将比对结果sample1.sam转为.bam格式 |
同样由于一批测序数据中有多个测序文件,因此需要循环以上过程,参考代码如下图所示:
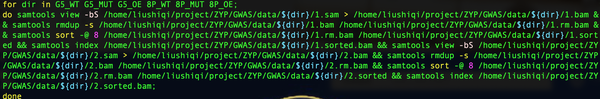
samtools去除PCR重复,排序和建立索引
得到基因组比对的去重复排序结果后,我们将比对信息中SNP的信息提取出来并整理成通用的突变格式VCF.
1 | SNP calling |
同样由于一批测序数据中有多个测序文件,因此需要循环以上过程,参考代码如下图所示:
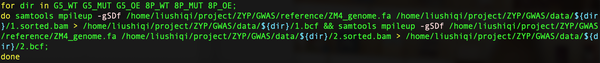
samtools进行SNP calling

bcftools进行vcf格式转换
得到的VCF文件记录了我们分析得到的外显子(对应WES)或基因组(对应WGS或WGRS)上的全部SNP信息,但是这里边有一些是低质量的噪音信息,我们需要通过设置一系列的筛选标准来将噪音去除,称为SNP质量过滤或数据清洗。我一般选用以下三个标准:
- –max-missing: 最大缺失值
- –minDP: 最小测序深度
- –minQ: 最小碱基质量
其中比较难以理解的是最大缺失值这个参数,vcftools官方给出的定义是这样的:

最大缺失值官方定义
我盯住的是0允许全部缺失而1代表不允许缺失,那就是说该值是介于0~1之间的浮点数,其大小代表着最少需要多少平行含有该SNP的比例。如0.3就代表至少30%的平行都有该SNP.
参考代码如下:
1 | vcftools --vcf sample1.vcf --minDP 3 --max-missing 0.3 --minQ 30 --recode --recode-INFO-all --out sample1.filtered |
同样由于一批测序数据中有多个测序文件,因此需要循环以上过程,参考代码如下图所示:
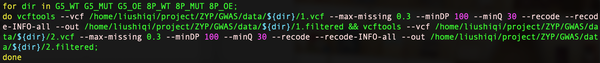
vcftools进行SNP质量过滤
利用前面下载的基因组和注释文件对质量过滤后的SNP进行注释, 【将基因组和注释文件放在同一路径下】!
1 | 我们下载的参考基因组注释文件是.gff格式的,需要转换为.gtf格式 |
1 | 建立注释索引 |
SNP注释结果文件预览结果如下图所示:

由左至右各列信息为:
- 所在行
- 同义/非同义突变
- 突变基因/外显子/核苷酸变化及位置/氨基酸变化及位置
- 染色体
- 突变起始位置
- 突变终止位置
- 原始序列
- 突变序列
- 同源/异源
- FQ: Phred probability of all samples being the same
- XXX
- 测序质量
同样由于一批测序数据中有多个测序文件,因此需要循环注释过程,参考代码如下图所示:
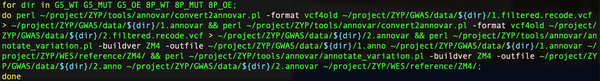
ANNOVAR进行SNP注释
可视化补充信息—-samtools统计测序覆盖度和深度:
前方步骤中得到的去除PCR重复排序后的比对结果sample1.sorted.bam, 可通过samtools统计其中包含的测序覆盖度和深度信息用于可视化,参考代码如下:
1 | samtools depth sample1.sorted.bam sample1.depth |
得到的.depth文件预览结果如下:
- 第一列为染色体名称
- 第二列为染色体上的核苷酸位置
- 第三列为该位置上的测序深度
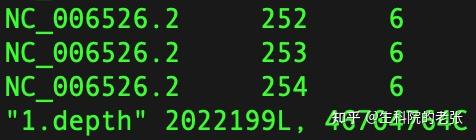
samtools进行测序覆盖度和深度统计
可视化:
将上一步得到的.depth文件用于统计测序深度与覆盖度,在Python中的参考代码如下:
1 | import seaborn as sns |
运行结果如下图所示:
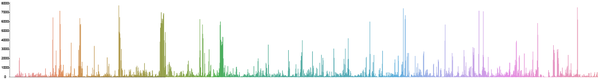
全基因组测序深度覆盖图
SNP在基因组浏览器IGV的可视化:
前方得到的去重复排序后的比对结果文件.sorted.bam可以在基因组浏览器IGV (下载链接为 http://software.broadinstitute.org/software/igv/download ) 中打开,从而在基因组全局和单核苷酸局部进行个性化浏览和分析。
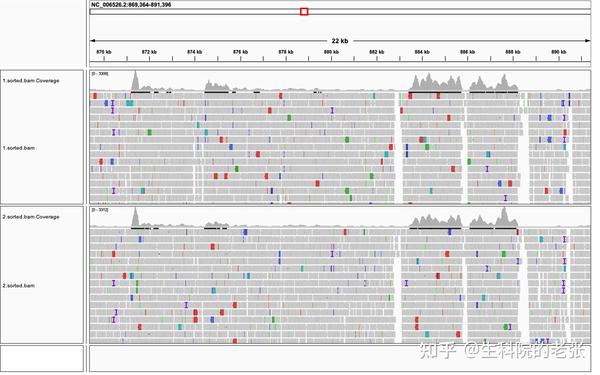
IGV可视化基因组
包括以下部分:
- SNV(单核苷酸变异,包括同义突变和非同义突变)
- Nonsynonymous SNV(非同义突变单核苷酸变异)
- Amino Acid Substitution(由非同义突变造成的氨基酸替换)
以ANNOVAR的SNP注释结果文件为输入,在Python中的参考代码如下:
1 | import pandas as pd |
如图所示,运行结束得到可视化结果和记录SNV与InDel的两个文件SNV.txt和InDel.txt .
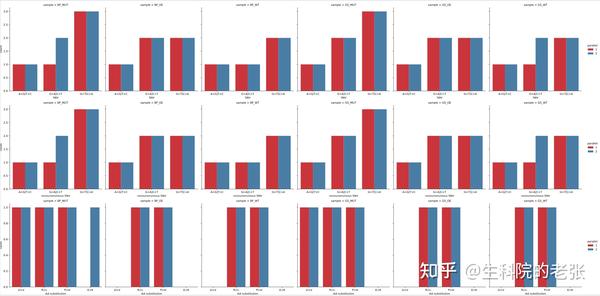
SNV and AA substitution
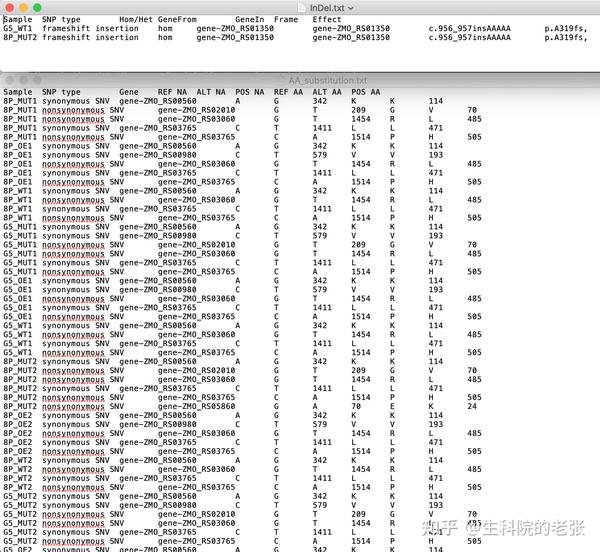
SNV and InDel
以前文中提到的测序深度文件.depth, ANNOVAR的SNP注释结果文件, 以及基因组注释文件.gff为输入,在R中的参考代码如下:
1 | library(stringr) #方便处理字符串 |
运行结果如图所示,由外向内依次是:
- 左上方图例和统计信息
- 基因组
- 染色体
- 测序深度与覆盖度
- 编码区和非编码区
- SNV和InDel密度
- SNV分布
- SNV类型
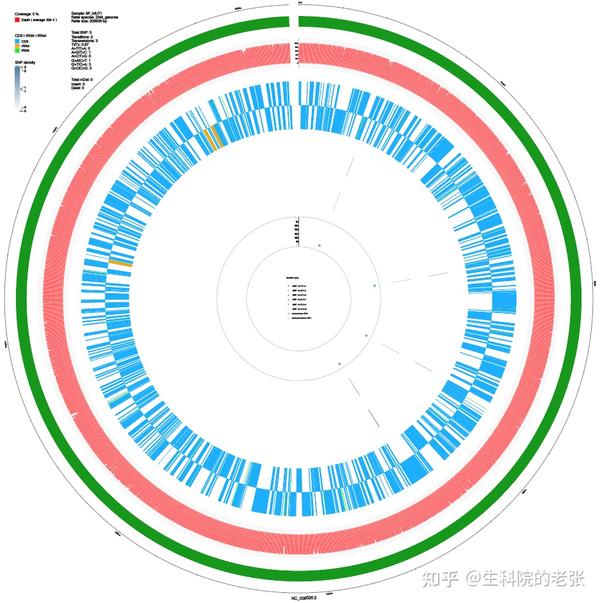
全基因组测序圈图
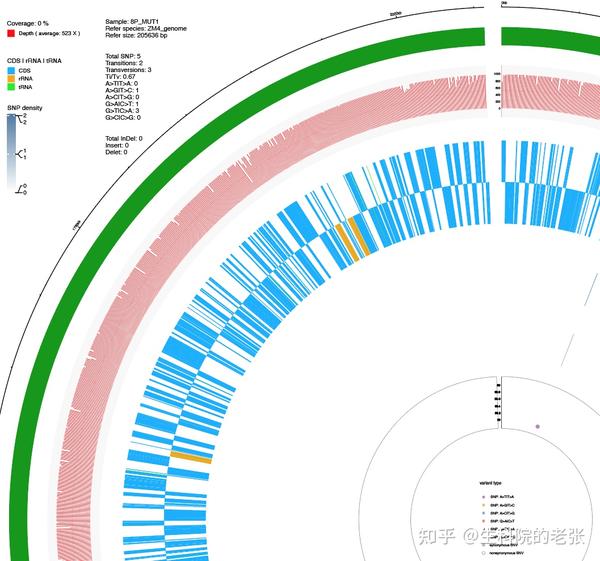
全基因组测序圈图参数
- 本文作者: Anderson
- 本文链接: http://nikolahuang.github.io/2024/03/17/WES数据分析流程-下/
- 版权声明: 转载请注明出处,谢谢。