

第一次发布: 2018年04月05日第8卷第7期 原文DOI: 10.21769/BioProtoc.2794
评审: David CisnerosSara E HowdenGanesh Swaminathan
cited from:Tidball, A. M., Swaminathan, P., Dang, L. T. and Parent, J. M. (2018). Generating Loss-of-function iPSC Lines with Combined CRISPR Indel Formation and Reprogramming from Human Fibroblasts. Bio-protocol 8(7): e2794. DOI: 10.21769/BioProtoc.2794.
简介
对于疾病和基础科学研究而言,功能丧失(LOF)突变是非常重要的。 在这里,研究人员提供了一个简单的方法来产生LOF iPSC系列,通过将CRISPR载体与附加型重编程质粒同时引入成纤维细胞,规避了传统基因编辑和已建立的iPSC系的克隆的技术挑战。 研究人员的实验已经产生了常染色体基因中所有3种基因型的几乎偶数。 此外,研究人员提供了一个详细的方法来维护和iPSC克隆的96孔板的基因分型。
背景
CRISPR / Cas9技术允许简单且特异地针对特定基因组位置进行基因编辑。将该技术与诱导性多能干细胞(iPSC)的疾病建模和再生医学潜力相结合将继续对生物医学研究产生前所未有的影响。然而,使CRISPR / Cas9系统适应iPSC已经提出了几个挑战。在细胞系中进行基因编辑的传统方法是用表达Cas9蛋白质的质粒和指导RNA(gRNA)转染细胞,然后产生单克隆并筛选所需的遗传改变。不幸的是,iPSC不适用于单细胞克隆。已经开发了几种补充媒介和克隆方法来克服这一困难,但仍然充满昂贵的设备(低氧培养箱),困难的技术步骤(FACS分选的单个iPSC的存活)或劳动密集型方案(亚克隆)(Forsyth ,2006; Miyaoka ,,2014)。此外,单细胞传代与iPSC中增加的基因组异常有关(Bai等人,2015)。已使用荧光或抗生素抗性遗传标记来克服克隆性问题和这些细胞中基因编辑的总体低效率,但需要通过设计有长同源臂(400-800bp)的靶向质粒对大盒进行同源重组, (Hendel等,,2014)。设计这些质粒需要很长时间。
为了克服这些障碍中的许多障碍,研究人员利用同时重编程和CRISPR / Cas9诱变的组合方法产生杂合和纯合丢失功能(LOF)iPSC系。 Howden及其同事首次提出了这种组合方法用于同源重组基因编辑(Howden等人,2015和2016),但是研究人员已经扩展并进一步定义了他们在最近的出版物(Tidball et。,2017)。该程序利用了iPSC重编程中固有的克隆步骤以及成纤维细胞培养物中更容易的转染(图1)。高效地产生大量具有所有三种基因型( 野生型/野生型,野生型/ indel和 indel / indel)的相似比例的克隆的能力将允许用于疾病建模的LOF iPSC系的快速发育和基础研究。
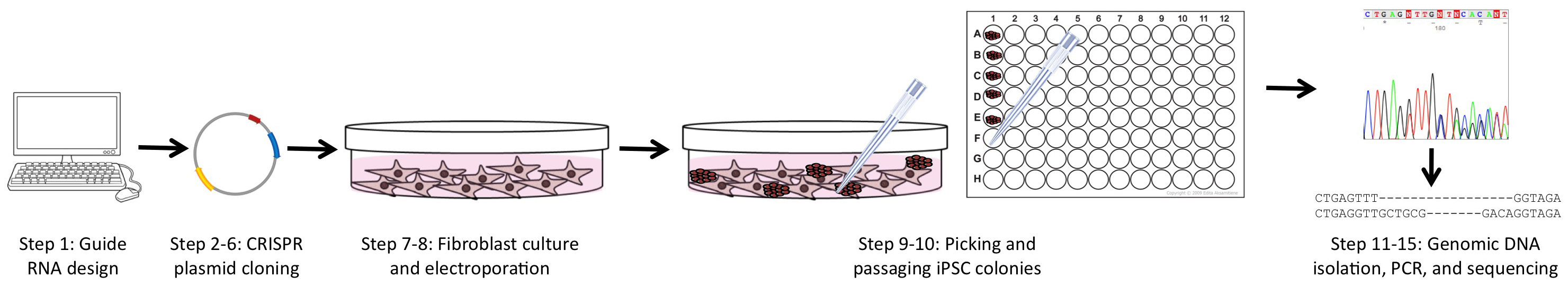
图1.实验工作流程概述本文中的方案分为4个主要部分,包括:指导RNA和PCR引物设计(步骤1),CRISPR质粒的产生和纯化(步骤2- 6),用CRISPR和重编程质粒(步骤7-8)进行成纤维细胞培养和电穿孔,并分离基因组DNA,然后对目标区域进行PCR并测序(步骤11-15)
材料和试剂
材料
- 1.5 ml Eppendorf管(VWR,目录号:89000-028)
- 10μl,200μl和1 ml移液枪头(USA Scientific,产品目录号:1111-3700,1111-0700和1111-2721)
- 用于细菌培养的14ml试管(Corning,Falcon ,产品目录号:352059)
- 锥形瓶(Corning,PYREX ®,目录号:4980-500)
- 10厘米组织培养皿(Corning,Falcon ,目录号:353003)
- 6孔板(Corning,Costar ®,产品目录号:3516)
- 5ml,10ml和25ml血清移液管(Aikali Scientific,ASI,目录号:SP205,SP210和SP225)
- 96孔板(Corning,Falcon ,目录号:353072)
- 无菌50毫升一次性移液器盆(Fisher Scientific,目录号:13-681-502)
- PCR管带(USA Scientific,目录号:1402-4700)
- 15 ml离心管(Fisher Scientific,目录号:05-539-12)
- Nalgene通用长期储存低温管(Thermo Fisher Scientific,Thermo Scientific TM,目录号:5000-1012)
细胞
人类包皮成纤维细胞(MTI-GlobalStem,目录号:GSC-3002)
试剂
- CRISPR质粒pX330-U6-Chmeric_BB-CBh-hSpCas9(Addgene,目录号:42230)
- 重编程质粒pCXLE-hOCT3 / 4-p53shRNA,pCXLE-hUL和pCXLE-hSK(Addgene,目录号:27077,27080,27078)
- 亚克隆效率DH5α感受态细胞(Thermo Fisher Scientific,Invitrogen TM,目录号:18265017)
- 与NEBuffer 2.1(New England Biolabs,目录号:R3539)一起提供的BbsI限制酶。
- QIAGEN PCR纯化试剂盒(QIAGEN,目录号:28104)
- 定制寡核苷酸(赛默飞世尔科技)
- T4 DNA连接酶试剂盒(Thermo Fisher Scientific,Invitrogen TM,目录号:15224017)
- SOC培养基(Thermo Fisher Scientific,Invitrogen TM,目录号:15544034)
- LB肉汤培养基(Thermo Fisher Scientific,Invitrogen TM,产品目录号:12780052)
- LB琼脂(Thermo Fisher Scientific,Invitrogen TM,产品目录号:22700025)
- 氨苄青霉素钠盐(Sigma-Aldrich,目录号:A9518-25G)
- QIAGEN Miniprep Kit(QIAGEN,目录号:27104)
- 甘油(Sigma-Aldrich,目录号:G5516-100ML)
- LKO.1 5’测序引物(5’-GACTATCATATGCTTACCGT-3’)
- 无内切QIAGEN Maxi Prep Kit(QIAGEN,产品目录号:12362)
- 磷酸盐缓冲盐水(PBS),pH7.4(Thermo Fisher Scientific,Gibco TM,产品目录号:10010023)
- 0.25%胰蛋白酶溶液(Thermo Fisher Scientific,Gibco TM,目录号:25200056)
- 生长因子耗尽基质胶(Corning,目录号:356230)
- 氖转染系统100μl试剂盒(Thermo Fisher Scientific,Invitrogen TM,目录号:MPK10025)
- TeSR-E7(STEMCELL Technologies,目录号:05910)
- mTeSR1(STEMCELL Technologies,目录号:85850)
- ROCK抑制剂Y-27632(Tocris Bioscience,目录号:1254)
- Accutase(Innovative Cell Technologies,目录号:AT104)
- ZR-96 Quick-gDNA Kit(Zymo Research,目录号:D3010)
- GoTaq绿色主混合物(Promega,目录号:M7122)
- 分子生物学级水(Thermo Fisher Scientific,Invitrogen TM,目录号:10977023)
- ZR-96 DNA测序清理试剂盒(Zymo Research,目录号:D4052)
- Gel 6x Loading Dye,Purple(New England Biolabs,目录号:B7024S)
- GeneMate LE快速溶解琼脂糖(BioExpress,GeneMate,目录号:E-3119-500)
- GelRed核酸凝胶染色10,000x在DMSO(Biotium,目录号:41002)
- Accugene 10 TBE缓冲液(Lonza,目录号:50843)
- Quant-IT PicoGreen dsDNA分析试剂盒(Thermo Fisher Scientific,Invitrogen TM,目录号:P7589)
- mFreSR(STEMCELL Technologies,目录号:05855)
- 液氮
- Dulbecco modified Eagle培养基(高葡萄糖)(Thermo Fisher Scientific,Gibco TM,目录号:11965118)
- 胎牛血清(FBS)(Thermo Fisher Scientific,Gibco TM,目录号:10437010)
- 青霉素 - 链霉素溶液(10,000U / ml)(Thermo Fisher Scientific,Gibco TM,目录号:15140122)
- MEM非必需氨基酸溶液100x(Thermo Fisher Scientific,Gibco TM,目录号:11140050)
- 成纤维细胞生长培养基(见食谱)
4.设备
- 便携式移液助剂(德拉蒙德,型号:DP-101,目录号:4-000-101)
- 层流罩(Thermo Fisher Scientific,Thermo Scientific TM,型号:1300系列II型A2型,目录号:1335)
- 氖电穿孔仪(Thermo Fisher Scientific,Invitrogen TM,目录号:MPK5000)
- Heraguard ECO Clean Bench(Thermo Fisher Scientific,Thermo Scientific TM,型号:Heraguard TM ECO,目录号:51029701)
- NanoDrop 2000分光光度计(Thermo Fisher Scientific,Thermo Scientific TM,型号:NanoDrop TM 2000)
- iCycler热循环仪(Bio-Rad Laboratories,型号:iCycler热循环仪)
- 水浴(Thermo Electron Corporation,产品目录号:51221073)
- 冰箱(Frigidaire,型号:FFTR2021QW1)
- 组织培养孵化器设置在37℃,5%CO 2(Thermo Fisher Scientific,Thermo Scientific TM,型号:Heracell TM 150i,目录号码:51026283)
- 带有平板接头的Thermo Scientific Sorvall St8 120v细胞培养离心机(Thermo Fisher Scientific,Thermo Scientific TM,型号:Sorvall TM St 8,120v)
- 明线血细胞计数器(Hausser Scientific,目录号:3110)
- 带8通道适配器的HandEvac手持式吸气器(Argos Technologies,产品目录号EV500)
- 奥林巴斯CKX41倒置细胞培养显微镜(奥林巴斯,型号:CKX41)
- 多道移液器(Thermo Fisher Scientific,Thermo Scientific TM,目录号:4662000和4662010)
- Sub-cell GT电泳细胞(Bio-Rad Laboratories,型号:Sub-Cell GT Cell)
- 凝胶成像仪
- Beckman Coulter DTX 880多模式检测器微孔板读数器(Beckman Coulter,型号:DTX 880)
- CoolCell LX冷冻容器(Biocision,目录号:BCS-405)
5. 程序
设计指南RNA(gRNA)和测序引物
- 使用基因组浏览器( genome.ucsc.edu ),该基因的所有转录本同种型中的第一个保守外显子(除非需要异构体特异性LOF突变)应该被鉴定并且序列被复制。然后可以将此序列插入指南RNA设计工具,例如 crispor.tefor.net 或 crispr.mit.edu 。
注:最重要的决定因素是特异性评分。尽可能使用最高分。研究人员之前报道的数据平均得分为88(Tidball等,2017)。记录最有可能脱离目标的网站以及可能筛选iPSC生产线中的脱靶插入。 crispr.mit.edu 未标识关闭 - 具有高度重复序列的目标(三重重复)。要注意的第二个分数是失分得分。这是一个计算出来的概率,任何给定的在切割位置上形成的indel将是不合格的,从而导致移码突变和功能丧失(推测)。研究人员之前报道的数据平均得分为63分。最后,效率得分应该> 50以确保很高的indel形成率。研究人员以前报告的数据平均得分为59分。 - 该gRNA可以作为标准脱盐寡核苷酸排序,序列CACCG-位于正向序列的5’端,而AAACC-位于反向序列的5’端。这些提供适当的突出端以插入px330(或相关)质粒以及gRNA开始时的G以启动从U6启动子的转录。
注意:不要将PAM序列(NGG)包含在gRNA寡核苷酸设计中。 - 如果可能的话,测序引物应该设计成具有〜200bp扩增子大小,几乎均匀地位于CRISPR切割位点的侧翼。 crispor.tefor.net 网站提供了一套很好的测序引物。使用NCBI引物blast site确认这些引物的解链温度和特异性( www .ncbi.nlm.nih.gov / tools / primer-blast / )(一定要使用人类基因组参考序列)。 
- 使用基因组浏览器( genome.ucsc.edu ),该基因的所有转录本同种型中的第一个保守外显子(除非需要异构体特异性LOF突变)应该被鉴定并且序列被复制。然后可以将此序列插入指南RNA设计工具,例如 crispor.tefor.net 或 crispr.mit.edu 。
载体质粒的限制性消化
- 将以下内容添加到1.5毫升管中:
2微克px330
5μl缓冲液2.1
2微升 Bbs I酶
水至50μl(首先加水至管) - 在37°C过夜消化
- 在第二天,将混合物在65°C加热灭活20分钟以停止消化反应。
- 使用QIAGEN PCR纯化试剂盒纯化切割的质粒。将纯化的质粒洗脱出水。
注意:PCR纯化试剂盒在分离切割的质粒时比凝胶提取更有效,并去除大部分小切片。第一次进行连接时,使用BbsI切割不含插入片段的质粒作为对照以确认菌落缺乏或几乎没有。切下的质粒可以在-20°C下保存数月。 - 使用NanoDrop分光光度计来确定切割质粒的浓度。
- 将以下内容添加到1.5毫升管中:
退火gRNA寡核苷酸以制备双链插入片段
- 将以下内容添加到1.5毫升管中:
1μlOligo1(100μM)
1μlOligo2(100μM)
7μl水
1μl缓冲液2 - 使用以下程序在PCR热循环仪中退火引物:
步骤1:95℃5分钟
步骤2:以-0.1°C /秒从95°C升温至25°C
步骤3:25℃30秒
第4步:保持在4°C。 - 在进行前,先将水稀释至1:250。
- 将以下内容添加到1.5毫升管中:
切割的载体质粒与退火的gRNA插入物的连接反应。
- 将以下组分添加到1.5毫升管中:
100ng来自步骤2的切割的px330质粒
2μl来自步骤3c的稀释寡聚双链体。
4μl的5x T4缓冲液
1μlT4连接酶
水至20μl。 - 在室温下孵育连接混合物2小时。
- 将以下组分添加到1.5毫升管中:
CRISPR-gRNA连接构建体的细菌转化
- 轻轻加入2μL连接反应到50μLDH5α热休克蛋白(冰上)。通过轻弹管子几次来混合。
- 将试管置于冰上30分钟。
- 将管子置于42°C水浴中20秒,热转换混合物
- 在冰上孵育2分钟。
- 加入950μL的SOC培养基,并将管放置在37℃的振荡器中,200rpm下1小时。
- 将40μl和400μl细胞铺在含有50μg/ ml氨苄青霉素的两个LB平板上。
- 在37°C孵育过夜
测序克隆
- 每个转化反应挑取3个克隆(使用P200移液管吸头)并接种到含有2ml含50μg/ ml氨苄青霉素的LB肉汤的14ml管中。
- 将含有孕育剂的14ml试管置于37℃的振荡器中,并使细菌培养物生长7小时(或直至混浊)。
- 根据制造商的说明使用QIAGEN Miniprep试剂盒从1.5 ml细菌中分离质粒。
- 将剩余的0.5ml细菌培养物与0.5ml 50%甘油混合并储存在-80℃冰箱中,直到获得测序结果。
- 使用NanoDrop分光光度计确定分离的质粒的浓度。浓度应该> 15 ng /μl以确保原料足够进行测序。
- 发送样品进行Sanger测序。通常,样品将在ddH 2 O中稀释至1ng /μl。使用1μM的LKO.1 5’测序引物(5’-GACTATCATATGCTTACCGT-3’)。
- 根据测序结果,挑选一个含有gRNA序列的克隆以建立最大培养。从小量制备培养物(步骤6d)接种0.1ml到锥形瓶中的100ml LB-Amp中,在37℃和200rpm的摇床上过夜培养。
- 根据制造商的说明,使用无内毒素的QIAGEN Maxi prep试剂盒从过夜细菌培养物中分离DNA。
- 使用NanoDrop确定质粒的浓度。
成纤维细胞培养
- 每隔一天更换成纤维细胞培养基(请参阅食谱)。
- 通过细胞大约每4天完全融合。
- 用10毫升1x PBS洗一次
- 每10厘米添加2毫升0.25%胰蛋白酶(10厘米的碟子)。
- 放入37°C培养箱中5分钟
- 一旦细胞分离,加入8ml成纤维细胞培养基,并将细胞转移到15ml锥形管中。
- 在室温下100g离心5分钟。
- 加入10ml新鲜的成纤维细胞培养基,并以1:3-1:6稀释倍数将细胞分裂到新的10cm培养皿中,并向每个培养板添加足够的额外培养基,总共10ml。
用CRISPR转染成纤维细胞并重编程质粒
- 转染前1-2小时,用在冰冷的DMEM / F12中稀释100μg/ ml的Matrigel包被6孔板。
每孔加入1 ml,37°C孵育 - 从亚汇合成纤维细胞中吸出培养基(铺满80%或更少)。
- 用10毫升1x PBS洗一次
- 加入2ml的0.25%胰蛋白酶并在37℃孵育5分钟。
- 将8ml成纤维细胞培养基添加到脱离的成纤维细胞中。
- 用5毫升血清移液管吸取几次上下,以确保即使是单细胞悬液。
- 使用血细胞计数器进行细胞计数。
- 将2.5×10 5个细胞转移到15ml锥形管中,并在室温下100×g离心5分钟。
- 吸取250μl缓冲液R到无菌1.5 ml试管中。加入三种重编程质粒各2.5μg和2.5μgCRISPR质粒。 (100μl的悬浮液用于电穿孔,50μl用于限制氖尖的气泡形成,如果发生电弧放电,100μl用于额外的电穿孔)。
- 重悬在含有重编程序列和CRISPR质粒的250μl缓冲液R中成纤维细胞沉淀。
- 在霓虹灯上使用100μl霓虹灯电穿孔电极,电压为1,650 V,10毫秒,3个脉冲。
- 如果电穿孔电弧(由闪光和爆裂声音表示),丢弃尖端的细胞悬液并用额外的150μl细胞悬液重复。
- 移液器成功从氖尖电穿孔细胞悬液到12毫升温暖的成纤维细胞培养基中。
- 从Matrigel包被的6孔板中取出Matrigel溶液,并向每个所需的孔中加入2 ml细胞悬液。
- 在37°C孵育板,18-24小时后更换成纤维细胞培养基。
- 电镀三天后,将培养基更换为TeSR-E7重新编程培养基。
- 每天更换TeSR-E7
- 在电穿孔后15-20天内出现可选择的集落(见图2)。
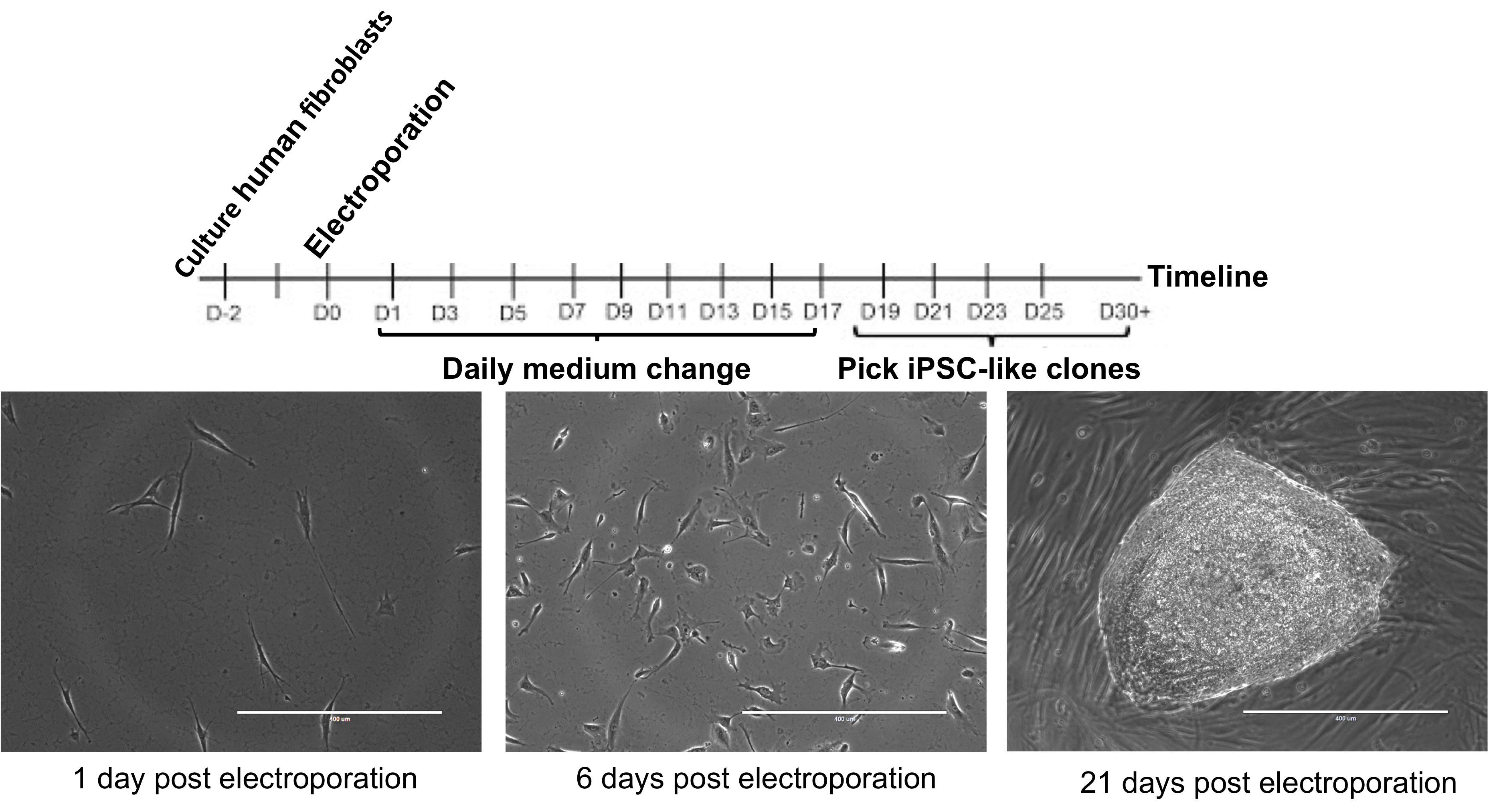
图2. iPSC重编程时间表。第21天的图像适合采摘,因为边界清晰,相对圆整,缺少附近的菌落。比例尺= 400微米。
- 转染前1-2小时,用在冰冷的DMEM / F12中稀释100μg/ ml的Matrigel包被6孔板。
挑选iPSC菌落
注意:典型的产量为每孔20-40个菌落。如果数量明显较低,这可能表明起始成纤维细胞培养物是不理想的(晚期传代,染色体异常等)。如果数量大得多,电穿孔过程中可能发生稀释错误,单克隆难以获得。
- 在采摘之前1-2小时,用在冰冷的DMEM / F12中稀释100μg/ ml的Matrigel涂布96孔板。每孔加入60μl,37°C孵育。
- 在挑选菌落之前,取出Matrigel溶液并加入100μl含10μMROCK抑制剂(Y-27632)的mTeSR1。
- 在含有倒置细胞培养显微镜(4x物镜)的HEPA工作站中,通过在微量移液器上用200μl移液器尖端进行解剖来挑选单个菌落。从皿中提取菌落后,用移液管以50μl体积移出菌落块(或多个),并放入96孔板的一个孔中。
注意:使用标准的200μl移液器吸头,凝胶加样吸头或类似小口径的吸头可能会过分剪切菌落。有关此技术的优秀视频,请访问: https://www.youtube.com/watch? v = B0lXzsHIcK8 。 - 偏向圆形分离的菌落挑选,以降低采集合并菌落导致异质基因型的风险。 (见图2)
注意:具有典型的重编程效率,可轻松从每个孔中挑选10-20个菌落。采摘可以连续进行;然而,不要在多天内从6孔板的相同孔中挑选,因为可能会从剩余的挑选菌落中形成次级菌落。 - 每天不用Y-27632将媒体更改为mTeSR1。
注意:步骤10-15使用8通道或12通道多通道移液器。
使用一次性移液器盆来容纳培养基和溶液以及收集废物。
传代iPSC克隆的96孔板
- 当大多数孔在100%或接近100%铺满时通过96孔板。
- 在每个新板上涂布60μl100μg/ ml基质胶,每孔用冰冷的DMEM / F12培养基稀释。在细胞培养箱中培养1-2小时。
- 使用8通道抽吸附件从96孔板吸取所有培养基。
- 用150μl1x PBS清洗平板一次
- 吸取并加入30μl室温Accutase到板的每个孔中。
在细胞培养箱中孵育5分钟 - 添加210微升含有10μM ROCK抑制剂,Y-27632的mTeSR1到每个孔中。
- 从以前的涂层板吸取Matrigel
- 将50μl细胞转移至每个待生成新平板的相应孔中。
- 转移细胞后,向每个孔中加入100μl额外的含10μMROCK抑制剂的mTeSR1以进一步稀释Accutase。
24小时后细胞汇合度应在10-30%之间
使用来自Zymo的ZR-96 Quick gDNA试剂盒从96孔板中分离基因组DNA
- 移出培养基,用100μlPBS洗一次。
- 加入300μl基因组裂解缓冲液并在室温下孵育10分钟。
- 将溶胞产物移至收集板上的Silicon-A Plate。
- 使用平板转接器,将平板在2,500×g 下离心5分钟。
注意:如果只使用1块平板,请务必正确平衡离心机。 - 向每个孔中加入200μlDNA预洗涤缓冲液并在2,500×gg下离心5分钟。丢弃流量。
- 向每个孔中加入300μlg-DNA洗涤缓冲液并在2,500×gg下离心5分钟。丢弃流量。
- 在2,500×g 下离心干燥5分钟以除去任何残留的缓冲液。
- 将硅A板放在洗脱板上。
- 加入40μlDNA洗脱缓冲液或水,并在室温下孵育5分钟。
- 2,500×g 离心5分钟
- 检查DNA是否由NanoDrop分光光度计纯化。
PCR来扩增目标区域
- 对于每个25μl反应混合物,将下列物质混合在1.5 ml Eppendorf管中:
12.5μlGoTaq 2x Master Mix
0.1μl100μM的每种引物
10.3μl分子生物学级水 - 在96孔PCR板或8孔PCR试管条的每个孔中加入23μl混合液进行PCR。
- 每个PCR孔加2μl模板
- 使用优化的PCR退火温度和标准GoTaq方案进行PCR。使用95℃2分钟变性步骤,然后进行以下40个循环:95℃30秒,优化的退火温度30秒和72℃30秒。最后,72℃5分钟的步骤将完成PCR产物的不完全延伸。
- 对于每个25μl反应混合物,将下列物质混合在1.5 ml Eppendorf管中:
用ZR-96 DNA测序纯化试剂盒(Zymo)纯化PCR产物
- 向PCR反应中加入240μl测序结合缓冲液。
- 将混合物转移到收集板上的Zymo-Spin IB-96板上。
- 2,500×g 离心5分钟
- 每孔加入300μl测序清洗缓冲液。
2,500×g 离心5分钟 - 每孔加入20μl水,并将平板固定在96孔PCR板上。
- 2,500×g 离心5分钟
- 在96孔板上纯化的PCR产物可以密封并保存在-20°C。
- 在含有1x GelRed核酸染色的2%琼脂糖凝胶上运行5μl与1μl6x上样染料混合的纯化PCR产物。通过在100V 1x TBE缓冲液中电泳1-2h分离条带,并使用凝胶成像仪检查正确的扩增子大小。 
使用PicoGreen
量化DNA浓度 注意:或者,使用NanoDrop分光光度计可以定量多个孔;然而,Zymo试剂盒具有高度230 nm峰的倾向,使DNA定量困难。
- 解冻Quant-IT PicoGreen dsDNA检测试剂盒组件。
- 加入1ml 20x TE缓冲液和19ml水,准备1x TE缓冲液。
- 在1x TE缓冲液(每孔50μl稀释的PicoGreen)中稀释PicoGreen 1:200。
- 通过在1x TE缓冲液中连续稀释DNA标准物至5,1,0.2,0.04和0ng /μl来制备一套DNA浓度标准品。
- 在一个清晰的96孔板中,加入50μl稀释的PicoGreen到15个孔中。
- 每个标准品加入50μl,一式三份。
- 5分钟后,使用荧光读板仪测量荧光,激发约480 nm,发射约520 nm。
- 通过在Excel中将标准DNA浓度和荧光强度输入到并列色谱柱中来制作标准曲线。生成XY散点图,右键单击数据点,然后选择“添加趋势线”。在“选项”标签中,选择“在图表上显示公式”。记录等式。
注意:如果图形不是线性的,则删除非线性区域中的数据点,并仅对线性区域中具有荧光读数的样本使用标准曲线方程。 - 加入50μl稀释的PicoGreen到96孔板的每个孔中。
- 每孔加入45μl1x TE
- 加入5μl来自步骤13的DNA。
- 阅读读板器,并使用步骤14h中生成的公式并将其乘以10来量化浓度。 
Sanger或下一代测序
- 现在可以对PCR产物进行测序。 Sanger测序是经济的;但是,插入错误会导致重叠的峰值。这些可用于使用像Mutation Surveyor( Softgenetics.com )这样的程序来识别突变,或手动识别突变形成。
- 较少劳动力密集型分析解决方案是使用Next-Gen测序。可以将样品提交给MGH(马萨诸塞州总医院)DNA核心进行CRISPR测序分析。简而言之,将条形码化的下一代测序适配器添加到每个样品的末端并复用。进行正向和反向100bp测序读取。然后将每个DNA种类定量为每个条形码总读数的百分比,并为每个样品列出具有百分比的比对序列。
建议的表征步骤
- 使用质粒DNA的qPCR测试附加型载体整合。
- G带核型分析或SNP芯片检测基因组异常。
- CRISPR脱靶PCR和测序以获得来自gRNA设计工具的最高脱靶位点评分。这可以用于外显子和内含子或仅外显子的脱靶。
- 使用myco特异性引物对细胞裂解液进行支原体污染检测。 
制作冷冻股票iPSC小瓶
注意:冷冻iPSC的高生命力最重要的方面是维持大的菌落。尽可能减少移液,并使用更大的刮刀来尽量减少菌落破裂。
- 从iPSC培养物中吸出培养基至1孔6孔板,约50%汇合(这比正常传代的最佳值高,但在冷冻时增加生存力)。
- 用2毫升37°C的DMEM / F12洗涤1次。
- 吸出,加入1毫升37°C的DMEM / F12,并加入1毫升冷的2U / ml Dispase溶液。
- 在37°C孵育8分钟
- 用2毫升37°C DMEM / F12吸取并清洗1次。
- 添加2毫升mTesr1培养基和废料
- 轻轻吸入15ml离心管中,并在15ml锥形管中以50gxg离心3分钟。
- 吸出培养基并加入1毫升mFreSR。
- 轻轻移入1.5 ml冷冻管中,置于CoolCell冷冻容器中,然后置于-80°C冷冻箱中过夜。
- 第二天将冷冻管转移到液氮储罐中。 
数据分析
Sanger测序分析
主要数据分析是分析测序数据并执行质量控制。首先,应排除PicoGreen DNA定量(低于检测限)的DNA浓度不足的样本。通常对这些样品进行测序将导致这些样品中出现奇数的indel识别(在其他克隆中未发现大的大量缺失)(数据未显示)。当使用Sanger测序时,也应排除具有3组重叠峰的样品,因为这些样品不是克隆的或可能是PCR之前DNA污染的结果。通过将重叠峰呼叫与indel起点下游的序列(其中峰开始重叠)(图3A)手动鉴定来自Sanger测序的indels。通过使用在线BLAST分析( https:// blast)可轻松识别出野生型或纯合的插入。 ncbi.nlm.nih.gov/Blast.cgi )。一定要使用人类基因组参考序列和“不连续的megablast”选项来允许indel对齐。杂合验证者可以通过Mutation Surveyor( Softgenetics.com )等计划来协助杂合子的识别,它根据野生型参考色谱仪并对齐以找到插入位置。不幸的是,这个程序对于复合杂合子插入不是很有效。通常,在Cas9切割位点附近形成插入缺失,其中最常见的是小缺失(表1)。在生成indels表时,使用Courier New。该字体对每个字符使用相同的大小,以便序列对齐。 
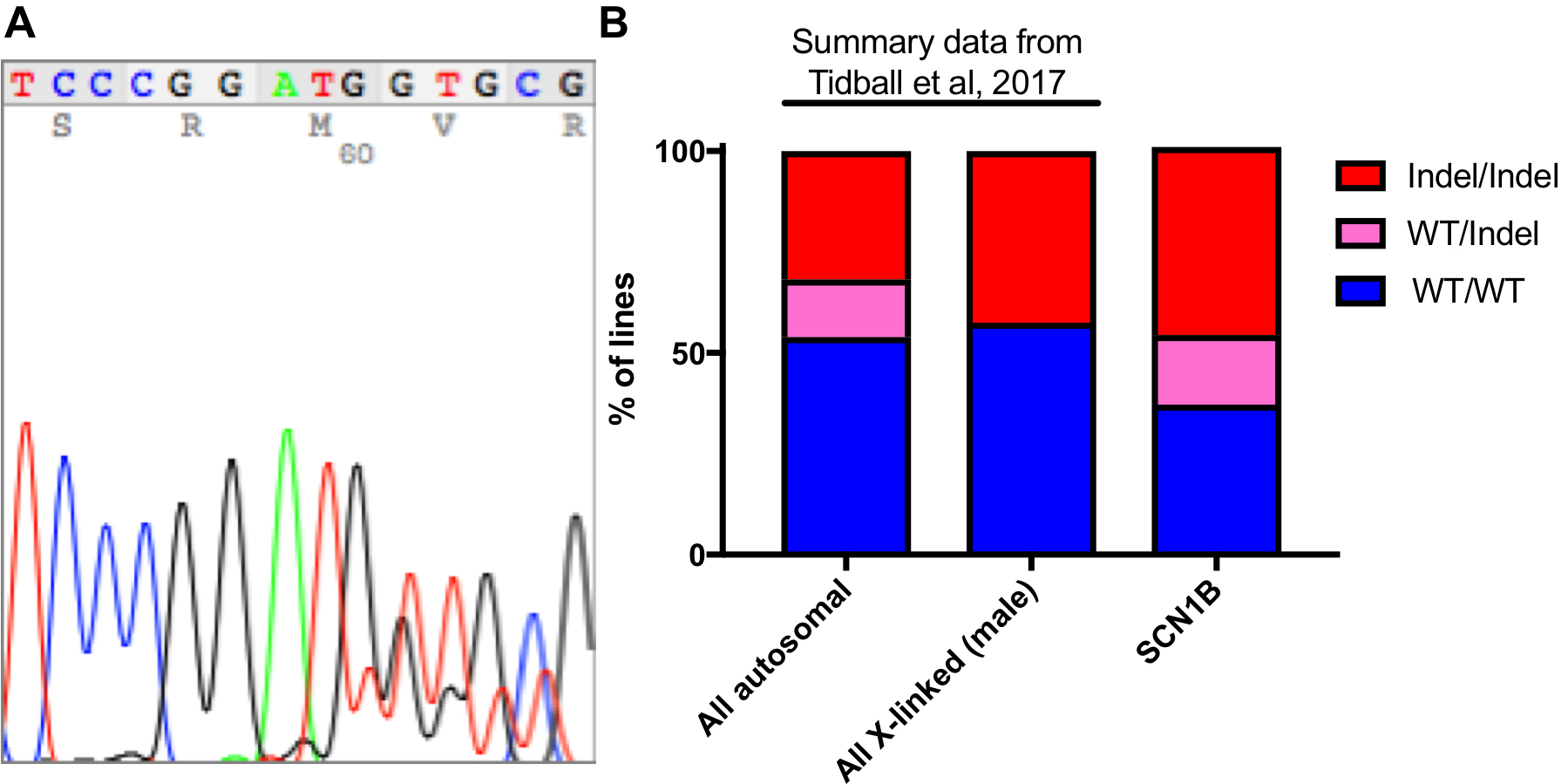
图3.典型的indel形态数据。 :一种典型的Sanger测序色谱图为杂合2 bp缺失。 B.来自Tidball 等的摘要数据。 (2017)与先前未发布的针对 SCN1B CRISPR / reprogramming的数据集相比较。这些数据包括22个WT / WT,10个WT / Indel和27个Indel / Indel线。
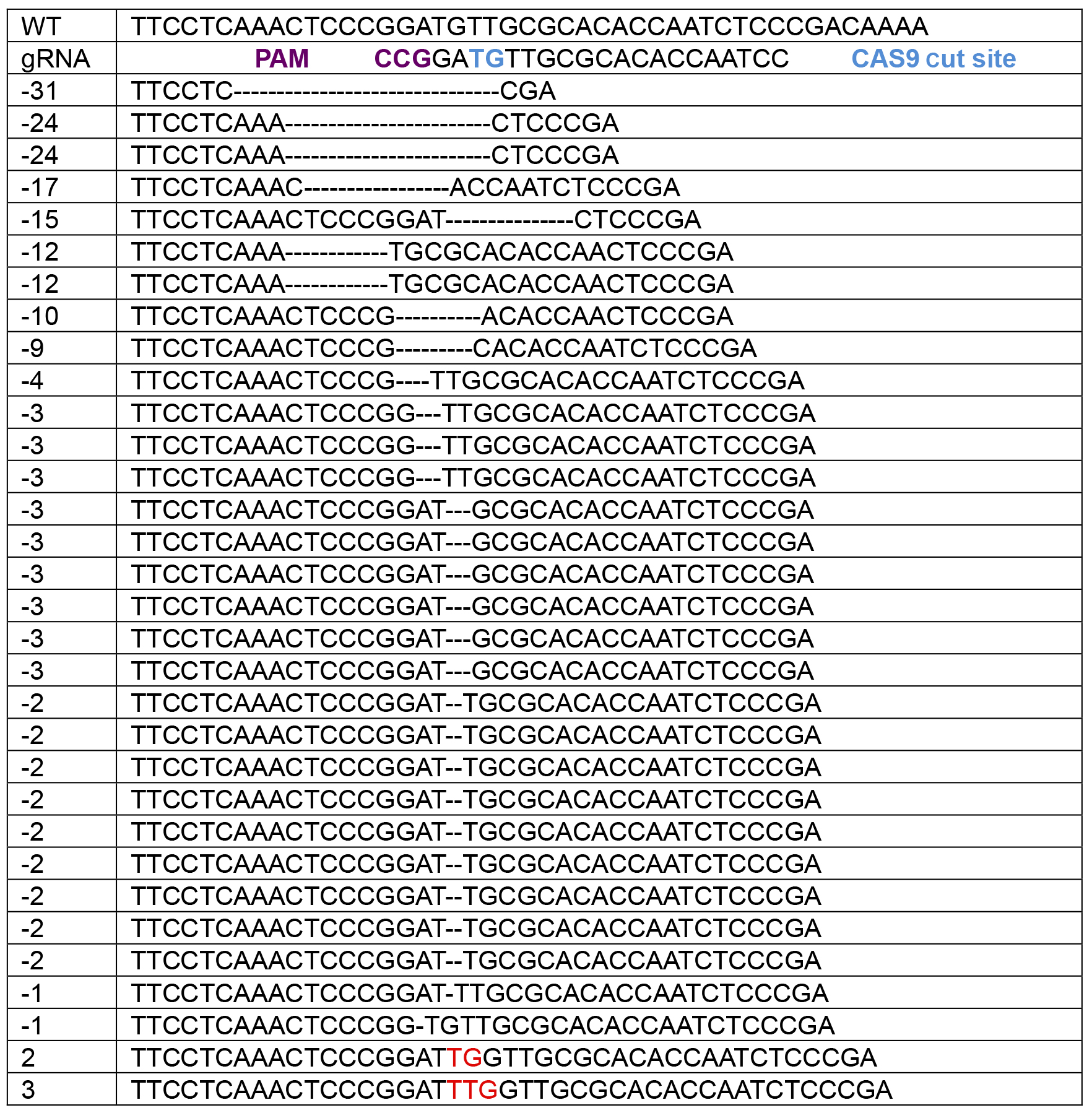
表1.通过具有>的单个实验形成的典型indels 50个克隆
NextGneration序列分析
NG测序将提供样品中发现的每种DNA物种的序列和读数的百分比。如果PCR不是特异性的,也可能存在一小部分脱靶DNA。除了indel鉴定,next-Gen测序的定量性质还允许鉴定异源系(2个或更多个克隆一起培育或挑选)。例如,如果杂合子线与50:50的比例发生偏离,则这可能表明异质性。请注意,大的插入可以稍微偏移该比例(例如59bp缺失杂合线导致几乎> 70%的WT量化)。
比较基因型的比例
尽管研究人员以前公布的5种癫痫基因的结果显示出所有三种基因型(Tidball等人,2017和图3B)的比例显着可重复性,但这可能会有所不同。如果鉴定出许多WT / indel系,但没有indel / indel系,则可能表明完全LOF导致无法复制或变得多能。为了确定这是否属实,应该将实验与另一个基因同时进行的实验进行比较。对于这样的实验,可以使用针对Tidball等人(2017)中突变的基因的指导RNA。可以使用Prism 7(GraphPad Software)中的卡方分析进行3种基因型比例的比较。
笔记
- 根据目标基因的不同,效率可能会偏离研究人员发表的内容。例如,当基因功能对于多能状态的增殖,存活或维持是必需的,因为LOF使得这些iPSC集落不太可能形成,所以可以预期完整的indel / indel LOF系列的数目较少。有趣的是,大量的克隆产生和研究人员的数据的可重复性确实允许基因之间的一些比较。例如,一个基因在每个indel / indel iPSC系中至少有一个框内突变。由于这与研究人员以前看到的以及预期的框架外突变百分比(CRISPOR.org)有很大差异,研究人员推测这个基因的表达对于iPSC形成是必需的。
- 在研究人员的实验中,研究人员一直小心地使用相同的质粒制剂和电穿孔试剂盒。如果对方案做出改变,导入的质粒数量可能会发生变化,从而导致每个基因型组的百分比发生变化。
培养基
成纤维细胞生长培养基
450毫升高糖DMEM
50毫升胎牛血清(FBS)
5毫升非必需氨基酸
2.5毫升Penn / Strep
Reference
- Bai, Q., Ramirez, J. M., Becker, F., Pantesco, V., Lavabre-Bertrand, T., Hovatta, O., Lemaitre, J. M., Pellestor, F. and De Vos, J. (2015). Temporal analysis of genome alterations induced by single-cell passaging in human embryonic stem cells. Stem Cells Dev 24(5): 653-662.
- Forsyth, N. R., Musio, A., Vezzoni, P., Simpson, A. H., Noble, B. S. and McWhir, J. (2006). Physiologic oxygen enhances human embryonic stem cell clonal recovery and reduces chromosomal abnormalities. Cloning Stem Cells 8(1): 16-23.
- Hendel, A., Kildebeck, E. J., Fine, E. J., Clark, J., Punjya, N., Sebastiano, V., Bao, G. and Porteus, M. H. (2014). Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep 7(1): 293-305.
- Howden, S. E., Maufort, J. P., Duffin, B. M., Elefanty, A. G., Stanley, E. G. and Thomson, J. A. (2015). Simultaneous reprogramming and gene correction of patient fibroblasts. Stem Cell Reports 5(6): 1109-1118.
- Howden, S. E., McColl, B., Glaser, A., Vadolas, J., Petrou, S., Little, M. H., Elefanty, A. G. and Stanley, E. G. (2016). A Cas9 variant for efficient generation of indel-free knockin or gene-corrected human pluripotent stem cells. Stem Cell Reports 7(3): 508-517.
- Miyaoka, Y., Chan, A. H., Judge, L. M., Yoo, J., Huang, M., Nguyen, T. D., Lizarraga, P. P., So, P. L. and Conklin, B. R. (2014). Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods 11(3): 291-293.
- Tidball, A. M., Dang, L. T., Glenn, T. W., Kilbane, E. G., Klarr, D. J., Margolis, J. L., Uhler, M. D. and Parent, J. M. (2017). Rapid generation of human genetic loss-of-function iPSC lines by simultaneous reprogramming and gene editing. Stem Cell Reports 9(3): 725-731.
- 本文作者: Anderson
- 本文链接: http://nikolahuang.github.io/2024/03/07/插入缺失的形成联合重编程生成功能丧失iPSC细胞系/
- 版权声明: 转载请注明出处,谢谢。